
Int J App Pharm, Vol 17, Issue 1, 2025, 465-473Original Article
FORMULATION AND PHARMACOKINETIC STUDY FOR LIQUISOLID COMPACTS OF CINACALCET HCL
M. SOMESU1, CHINAM NIRANJAN PATRA2*, GOUTAM KUMAR JENA2, 2DIPTHI SHREE, 3SUDARSAN BISWAL
1College of Pharmaceutical Sciences, Berhampur, Affiliated to Biju Patnaik University of Technology, Rourkela, India. 2Roland Institute of Pharmaceutical Sciences, Berhampur, India. 3Directorate of Drugs Controller, Health and Family Welfare Department, Govt. of Odisha, Bhubaneswar, India
*Corresponding author: Chinam Niranjan Patra; *Email: drniranjanrips@gmail.com
Received: 08 Aug 2024, Revised and Accepted: 09 Oct 2024
ABSTRACT
Objective: The objective of this study is to enhance the flowability, compressibility, and oral bioavailability of Cinacalcet Hydrochloride (HCl) using the liquisolid technique. It is a calcimimetic drug approved for treating secondary hyperparathyroidism in chronic kidney disease patients faces challenges due to its poor aqueous solubility and low bioavailability (20-25 %).
Methods: To address this, we formulated cinacalcet HCl liquisolid compacts with tween 80 and labrasol as the non-volatile solvents, neusilin US2 as the carrier material, and aerosil as the coating material. Our comprehensive analysis included Fourier-transform Infrared Spectroscopy (FT-IR), Differential Scanning Calorimetry (DSC), Powder X-ray diffraction (P-XRD), kawakita analysis and heckel analysis, quality control tests and pharmacokinetic study.
Results: The liquisolid powders of cinacalcet HCl exhibited desirable flowability and compressibility for processing into a tablet dosage form. Kawakita and Heckel analysis revealed reduced cohesiveness and increased plasticity. FT-IR and DSC studies did not exhibit any interaction between drug and carriers. P-XRD study for liquisolid formulation did not exhibit any peaks due to the presence of cinacalcet HCl in molecular form. In vitro dissolution study revealed 37 times improvement in dissolution at 30 min. The Area Under the Curve (AUC) values showed a 2.5-fold increase in oral bioavailability.
Conclusion: Overall, the liquisolid approach promises to develop a stable and scalable solid dosage form with improved flowability, compressibility, and oral bioavailability.
Keywords: Kawakita analysis. Heckle analysis, Dissolution rate, and Pharmacokinetic study
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i1.52296 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Poorly water-soluble drugs exhibit slow dissolution rates, which pose a significant challenge in formulating oral pharmaceutical dosage forms [1]. Enhancing solubility and dissolution rate is crucial for improving drug absorption along the intestinal tract [2]. Various approaches have been explored to address this issue, including solid dispersions [3, 4], inclusion complexation [5, 6], spray-drying techniques [7], lyophilization [8], micronization [9], and microwave [10] for Improving the rate of dissolution.
The liquisolid approach, devised by Spireas and their team, offers a strategy to increase the dissolution rate of poorly water-soluble pharmaceuticals [11]. The technique involves incorporating poorly water-soluble pharmaceuticals into a non-volatile solvent that is miscible with water, either in dissolved or suspended form. This mixture can then be transformed into a flowable, compressible, non-adherent powder [12]. The process involves using a porous carrier and a coating material. A dry powder is produced by covering the wet porous coating material with fine particles [13]. The technique is versatile and can be applied to a wide range of drugs, including those with poor aqueous solubility. It is also adaptable to various dosage forms, such as tablets and capsules [14].
Cinacalcet HCl, a calcimimetic medication, has received approval for treating secondary hyperparathyroidism in individuals undergoing dialysis for chronic renal disorder [15]. Moreover, cinacalcet HCl is employed to manage hypocalcemia in patients with parathyroid cancer. The primary obstacles in developing effective formulations for this compound are its limited aqueous solubility and low bioavailability, reported to be between 20 and 25% [16]. This study aims to enhance flowability, compressibility, dissolution properties, and oral bioavailability using the liquisolid technique.
MATERIALS AND METHODS
A gift sample of Cinacalcet HCl was obtained from Dr. Reddy’s Laboratories (DRL), Hyderabad, India. A gift sample of Neusilin US2 was obtained from Fuji Chemical Industries Co Ltd. Mumbai, India. Labrasol came as a gift sample from Gattefosse India Ltd. Microcrystalline Cellulose (MCC) and tween 80 were procured from Himedia, India.
Pre-formulation study
Solubility study
Following the standard protocol, the saturation solubility of cinacalcet HCl was examined in a range of non-volatile liquid vehicles, as shown in fig. 1. These non-volatile liquids were saturated with cinacalcet HCl, and then stirred for 48 h at 25 °C. The solutions were then filtered, centrifuged, and subjected to Ultraviolet (UV) Visible spectrophotometric examination at 279 nm.
Determination of loading factor
To determine the maximum liquid load capacity, three different porous carriers, namely neusillin US2, sylysia 730, fujicalin were selected and mixed with one common coating material i. e., aerosil at an R-value of 20. Admixtures of liquid powder were made at R = 20. To these admixtures of porous carrier and coating material (10 g), increasing amounts of non-volatile liquid (Tween 80 and labrasol) were added. After mixing for three minutes, the mixture was kept overnight and the angle of repose was determined. The admixture showing the angle of repose 25 was selected [17].
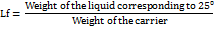
Preparation of liquisolid tablet
Two non-volatile solvents, namely Tween 80 and Labrasol, were chosen for preparing liquisolid compacts due to their higher solubilization capacity to dissolve cinacalcet HCl. The drug was dissolved separately in these selected solvents and vortexed for 10 min (table 1). Subsequently, the necessary quantity of Neusilin US2 and aerosol (20:1 ratio) was incorporated as carrier and coating agents. Cross-povidone and talc were subsequently introduced and mixed for ten minutes until a free-flowing powder was achieved. This liquisolid powder was then directly compressed into tablet form using a Minipress-II, Karnavati, Ahmedabad.
Characterization
Flowability
The flow properties of all formulations (F1 to F12), such as Carr's index, angle of repose, and Hausner’s ratio were evaluated as per standard protocol [18].
Kawakita analysis
A 50 ml glass measuring cylinder was taken and filled with 5 g of cinacalcet HCl and 5 g of cinacalcet HCl-loaded liquisolid formulation. The Initial bulk volume was designated as V0 and the tapped volume after N number of tappings was designated as V [19].
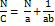
Where a is compatibility, and 1/b is cohesiveness. The C is the degree of volume reduction, V0 is the original volume and V is the tapped volume as follows:
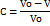
Graphical representation of N/C against the number of taps (N) yielded a linear relationship, from which the numerical values of constants a and 1/b were determined. (N = 0, 50, 100, 150, and 200).
Heckel analysis
Tablets were produced using a hydraulic pellet press (Model KP, M/S Kimaya Engineers Pvt Ltd, Mumbai, India) equipped with 12 mm flat-faced punches. A range of compression pressures (12.5, 25, 37.5, and 50 kg/cm²) were applied to both the drug and its liquisolid formulations. The dimension of each compact was measured using a digital slide caliper (Mitutoyo Co., Kawasaki, Japan) and weighed precisely (n = 10). The diameter, thickness, and weight of each compact were measured. The compaction characteristics of each compact were calculated using the Heckel equation [20]. The Heckel equation is described as follows.
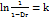 P+A
P+A
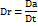
In this context, Dr, Da, and Dt represent the relative, true, and apparent densities, respectively, at the applied pressure (P). The slope (K) corresponds to the inverse of the material’s yield pressure (Py), while A is the intercept determined by the compact volume.
Table 1: Formulation of liquisolid compacts
| Formulation code | Cinacalcet HCl (mg) |
Tween 80 (ml) |
Tween 80 (mg) |
Weight of liquid medication (mg) W | R | Lf | Quantity of carrier Q in mg (Q=W/lf) | Quantity of coating (q) in mg q = Q/R | Cross povidone (mg) |
Talc (mg) |
Tablet weight (mg) |
| F1 | 30 | 0.6 | 460 | 490 | 20 | 3.29 | 149 | 7.5 | 0 | 4.5 | 651 |
| F2 | 30 | 0.6 | 460 | 490 | 20 | 3.29 | 149 | 7.5 | 2 | 4.5 | 653 |
| F3 | 30 | 0.6 | 460 | 490 | 20 | 3.29 | 149 | 7.5 | 4 | 4.5 | 655 |
| F4 | 30 | 0.6 | 460 | 490 | 20 | 3.29 | 149 | 7.5 | 6 | 4.5 | 657 |
| F5 | 30 | 0.6 | 460 | 490 | 20 | 3.29 | 149 | 7.5 | 8 | 4.5 | 659 |
| F6 | 30 | 0.6 | 460 | 490 | 20 | 3.29 | 149 | 7.5 | 10 | 4.5 | 661 |
| Formulation code | Cinacalcet HCl (mg) |
Labrasol (ml) |
Labrasol (mg) |
Weight of liquid medication (mg)W | R | Lf | Quantity of carrier Q in mg (Q=W/lf) | Quantity of Coating (q) in mg q = Q/R | Cross povidone (mg) |
Talc (mg) |
Tablet weight (mg) |
| F7 | 30 | 0.7 | 505 | 535 | 20 | 3.12 | 171 | 8.5 | 0 | 4.5 | 719 |
| F8 | 30 | 0.7 | 505 | 535 | 20 | 3.12 | 171 | 8.5 | 2 | 4.5 | 721 |
| F9 | 30 | 0.7 | 505 | 535 | 20 | 3.12 | 171 | 8.5 | 4 | 4.5 | 723 |
| F10 | 30 | 0.7 | 505 | 535 | 20 | 3.12 | 171 | 8.5 | 6 | 4.5 | 725 |
| F11 | 30 | 0.7 | 505 | 535 | 20 | 3.12 | 171 | 8.5 | 8 | 4.5 | 727 |
| F12 | 30 | 0.7 | 505 | 535 | 20 | 3.12 | 171 | 8.5 | 10 | 4.5 | 729 |
Quality control tests
The prepared liquisolid tablets were subjected to quality checks which included drug content, hardness, disintegration time, weight variation, and friability test was performed as per standard protocol [21].
In vitro dissolution test
Liquisolid formulations exhibiting disintegration time (<10 min) were chosen for an in vitro dissolution study (F5, F6, F11 and F12). Additionally, the cinacalcet HCl underwent dissolution testing. The study employed United States Pharmacopeia (USP) type II paddle equipment operating at 50 rpm, using 0.1 N HCl as the dissolution medium for a span of 2 h. The dissolution profiles were analyzed to determine key parameters such as dissolution efficiency, average dissolution time, and the applicability of the Hixson-Crowell cube root model [22].
Fourier transform infrared (FT-IR) study
Approximately two milligrams of the sample were thoroughly mixed with a predetermined amount of dry potassium bromide powder and subsequently compressed into disks. Fourier-transform infrared spectroscopy analysis of cinacalcet HCl and the chosen liquisolid formulations (F5 and F12) adhered to standard operational procedures.
Differential scanning calorimetry (DSC study)
Accurately weighed samples of cinacalcet HCl, F5, and F12 were enclosed in sealed aluminum pans and subjected to thermal analysis. (Heating rate 10 °C/min from ambient temperature to 220 °C).
Powder X-ray diffraction (P-XRD) study
Powder X-ray diffraction patterns were obtained for both pure cinacalcet HCl and liquisolid tablets (F5 and F12) within the 2 to 70° 2θ angular range.
Stability study
Stability studies were conducted on formulation F5 for six months under accelerated conditions (40±2 °C/75±5% RH) in compliance with International Council for Harmonization (ICH) Q1A R2 guidelines. The formulation was assessed for drug content, disintegration time, and dissolution at the 30-minute time point [23].
Pharmacokinetic study
Twelve male albino rabbits with a body weight of 2 kg were carefully selected for this study. Group 1 (six rabbits) received the cinacalcet HCl liquisolid tablet (F5) as the test substance, while Group 2 (six rabbits) was administered the standard aqueous suspension of cinacalcet HCl with approval no 95 of Institutional Animal Ethical Committee (IAEC) of Roland Institute of Pharmaceutical Sciences (RIPS) (Regd. no. 926/PO/Re/S/06/CPCSEA). The albino rabbits were procured from Shah Enterprise, Kolkata, West Bengal, bearing registration number 1828/PO/Bt/S/15/CPCSEA. The animals were acclimatized in the animal house of RIPS with air conditioned temperature of 25 C. The dose for rabbits was calculated as 2.3 mg. A 2.3 mg dose was given orally using a Ryle’s tube. Blood samples (0.5 ml) were drawn from the marginal ear vein of male rabbits at specific time points (0, 0.5, 2, 6, 12, 24, and 48 h) with a 24-gauge needle and collected in Eppendorf tubes. Pharmacokinetic parameters, including Maximum Plasma Drug Concentration (Cmax), Time to maximum Drug Concentration (Tmax), and AUC were calculated. Sample analysis followed a previously published Ultra-fast Liquid Chromatography (UFLC) method for quantifying the compound. Chromatographic separation was achieved on a C18 column using a mobile phase consisting of acetonitrile and Tert-butyldimethylsilyl chloride (TBHS0 (50:50) delivered at a flow rate of 1 ml/min [24].
RESULTS AND DISCUSSION
Solubility study
Cinacalcet HCl was most soluble in tween 80, reaching a concentration of 62.3 mg/ml, whereas labrasol achieved a solubility of 43.1 mg/ml (fig. 1). Tween 80 and labrasol were selected for further studies. Both tween 80 and labrasol enhance solubility by forming micelles or emulsions. These non-volatile solvents can encapsulate lipophilic drugs within their micellar structures, effectively increasing their solubility in aqueous media [25].
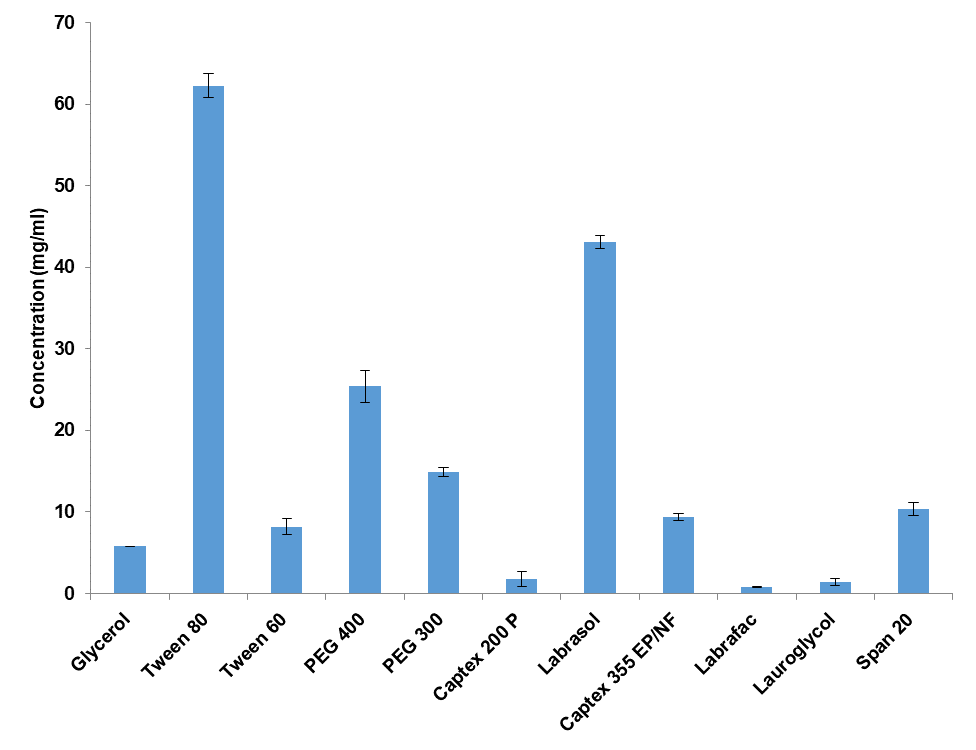
Fig. 1: Saturation solubility of cinacalcet HCl in non-volatile solvents. Data expresses as mean±SD, n = 6
Liquid loading factor
The flowable liquid retention potential values for admixture of Neusilin US2 and aerosol (R = 20) were 3.09% (w/w) and 3.12 % (w/w) for tween 80 and labrasol, respectively, at a carrier-to-coating ratio of R = 20. The flowable liquid retention potential values for sylysia and fujacalin were very low compared to neusillin US2. For further studies, Neusillin US2 was selected as a porous carrier. The porous structure of Neusillin US2 creates numerous adsorption sites, enhancing its capacity to bind substances [26]. Neusillin US2 has been used successfully in converting liquid formulations like solid lipid nanoparticles, nanostructured lipid carriers, and self-emulsifying drug delivery systems into free-flowing powders with desirable flowability and compressibility [27, 28].
Preparation of liquisolid compact
Liquisolid powders were formulated using a mixing technique. The chosen method is scalable and adaptable. Cinacalcet HCl exhibited complete solubility in both tween 80 and labrasol. An increase in Neusilin US2 content within the liquisolid formulation resulted in improved flowability, thereby enhancing its suitability for tablet compression.
Flowability
Flowability assessments indicated that pure cinacalcet HCl exhibited suboptimal flow properties. Conversely, all liquisolid formulations demonstrated favorable tableting characteristics despite variations in non-volatile solvents (table 2). The improved flowability of the liquisolid formulations is likely due to the adsorptive and compressible properties inherent to Neusilin US2. Additionally, the presence of an aerosol coating on the wet surface of the porous carrier contributes to improved flowability [29].
Kawakita analysis
Liquisolid formulations demonstrated superior flowability compared to cinacalcet HCl, as indicated by lower 'a' values, a measure of compatibility. Additionally, these formulations exhibited reduced cohesiveness, as evidenced by lower '1/b' values, when compared to cinacalcet HCl (table 3).
Heckel analysis
The intercept value 'A' for liquisolid formulations was comparable, yet notably greater than that of cinacalcet HCl. The elevated K value observed in liquisolid formulations suggests enhanced compressibility and a higher degree of plastic deformation during compaction (table 4).
Table 2: Flowability of liquisolid formulations
| Formulation | Angle of repose (˚) | Carr’s index (%) | Hausner’s ratio |
| Cinacalcet HCl | 41.3±3.1 | 29.6±2.43 | 1.9±0.06 |
| F1 | 24.5±1.2 | 17.4±0.9 | 1.14±0.05 |
| F2 | 21.2±1.15 | 16.1±0.8 | 1.11±0.06 |
| F3 | 20.6±1.2 | 18.8±1.2 | 1.21±0.03 |
| F4 | 21.7±1 | 16.4± 0.5 | 1.21±0.07 |
| F5 | 23.6±1.25 | 19.3± 0.4 | 1.18±0.02 |
| F6 | 23.5±1.2 | 18.3± 0.9 | 1.23±0.03 |
| F7 | 24.2±1.15 | 16.6± 0.7 | 1.21±0.07 |
| F8 | 20.1±1.1 | 17.1± 0.8 | 1.24±0.06 |
| F9 | 21.6±1.25 | 14.8±0.85 | 1.21±0.02 |
| F10 | 23.4±1.2 | 16.6± 0.75 | 1.23±0.04 |
| F11 | 24.4±1.1 | 15.5± 0.8 | 1.24±0.06 |
| F12 | 23±1.15 | 16.7± 0.9 | 1.20±0.05 |
Data expresses as mean±SD, n = 6.
Table 3: Kawakita analysis of liquisolid formulation
| Formulation | Compatibility (a) | Cohesiveness (1/b) | Coefficient of determination (r2) |
| Cinacalcet HCl | 0.64 | 25.43 | 0.997 |
| F1 | 0.16 | 11.52 | 0.951 |
| F2 | 0.18 | 18.31 | 0.952 |
| F3 | 0.16 | 17.25 | 0.978 |
| F4 | 0.15 | 14.25 | 0.956 |
| F5 | 0.18 | 20.14 | 0.953 |
| F6 | 0.19 | 18.23 | 0.998 |
| F7 | 0.17 | 15.85 | 0.996 |
| F8 | 0.16 | 21.34 | 0.992 |
| F9 | 0.12 | 17.55 | 0.994 |
| F10 | 0.18 | 14.16 | 0.997 |
| F11 | 0.19 | 16.09 | 0.995 |
| F12 | 0.17 | 18.15 | 0.991 |
Table 4: Heckel analysis of liquisolid formulation
| Formulations | Slope (K) | Intercept (A) | Yield pressure (P) | Coefficient of determination (r2) |
| Pure drug CINH | 0.002 | 0.356 | 1125 | 0.935 |
| F1 | 0.007 | 0.853 | 200 | 0.972 |
| F2 | 0.006 | 0.842 | 275 | 0.975 |
| F3 | 0.004 | 0.836 | 250 | 0.959 |
| F4 | 0.005 | 0.785 | 256 | 0.969 |
| F5 | 0.004 | 0.815 | 289 | 0.987 |
| F6 | 0.006 | 0.729 | 310 | 0.968 |
| F7 | 0.005 | 0.761 | 249 | 0.973 |
| F8 | 0.006 | 0.783 | 287 | 0.951 |
| F9 | 0.007 | 0.827 | 276 | 0.976 |
| F10 | 0.008 | 0.835 | 263 | 0.983 |
| F11 | 0.005 | 0.796 | 284 | 0.964 |
| F12 | 0.006 | 0.829 | 273 | 0.967 |
Table 5: QC tests for liquisolid tablets
| Formulations | Drug content* (%) | Weight variation** (mg) | Friability** (%) | Hardness*** (Kg/cm2) | Disintegration time*** (min) |
| F1 | 98.4±1.8 | 651.3±15.2 | 0.5±0.05 | 5.6±0.175 | 33.6±1.6 |
| F2 | 99.5±3.2 | 653.4±15.5 | 0.8±0.04 | 5.5 ±0.185 | 16.5 ±1.3 |
| F3 | 97.6±2.9 | 655.3±19.6 | 0.6±0.07 | 5.8 ±0.19 | 14.1 ±1.2 |
| F4 | 98.4±2.5 | 657.3±14.1 | 0.3±0.06 | 5.7 ±0.185 | 11.6 ±1.05 |
| F5 | 96.3±1.7 | 659.3±8.4 | 0.7±0.04 | 5.3 ±0.255 | 4.8 ±1.5 |
| F6 | 95.2±4.8 | 661.3±17.2 | 0.6±0.08 | 5.7 ±0.26 | 4.1 ±0.95 |
| F7 | 99.6±3.6 | 719.5±18.2 | 0.3 ±0.06 | 5.3 ±0.245 | 25.3 ±0.8 |
| F8 | 98.1±2.1 | 721.7±15.4 | 0.4 ±0.02 | 5.5 ±0.26 | 18.3 ±0.6 |
| F9 | 96.6±1.3 | 723.5±17.6 | 0.5±0.01 | 5.6 ±0.29 | 19.6 ±1.51 |
| F10 | 98.8±2.8 | 725.8±18.8 | 0.8 ±0.07 | 5.3 ±0.295 | 16.3±0.7 |
| F11 | 97.3±1.8 | 727.8±9.2 | 0.2 ±0.05 | 5.6.±0.3 | 8.6±0.35 |
| F12 | 98.3±1.5 | 729.4±12.5 | 0.3 ±0.04 | 5.5 ±0.31 | 7.5±0.15 |
Data expresses as mean±SD. *n = 10, **n = 20, ***n = 6.
Quality control tests for liquisolid tablets
All liquisolid tablets demonstrated drug content surpassing 95 %, confirming the homogeneous mixing of the drug within the excipient matrix. All formulations exhibited weight variations that complied with the ±5% acceptance criterion, indicative of satisfactory flow properties. Formulations F1 to F4 and F7 to F10 were rejected because they took more time to disintegrate, which may be ascribed to the lower proportion of disintegrating agent. Following the successful completion of tablet quality control assessments, liquisolid formulations F5, F6, F11, and F12 were selected for the next phase of the study (table 5).
In vitro dissolution test
In the dissolution study, pure drug cinacalcet HCl and liquisolid tablets F5, F6, F11, and F12 underwent a 2 h test in 0.1 N HCl (fig. 2). Interestingly, less than 15% of cinacalcet HCl dissolved during 2 h of dissolution study. However, liquisolid tablets with tween 80 as a non-volatile solvent (F5 and F6) exhibited nearly 100% dissolution of cinacalcet HCl in 60 min, whereas liquisolid tablets with labrasol as a non-volatile solvent (F11 and F12) demonstrated 100 % dissolution in 80 min. The higher rate of dissolution for F5 and F6 can be ascribed to the higher solubility of cinacalcet HCl in tween 80. Significantly, formulations F5, F6, F11, and F12 exhibited dissolution rates that were 37, 37, 25, and 28 times higher, respectively, compared to that of cinacalcet HCl when assessed using the Q30 parameter (table 6). Liquisolid tablet F5 exhibited the shortest mean Dissolution time (MDT) among all formulations tested, indicating enhanced dissolution efficacy relative to pure cinacalcet HCl [30]. Similar improvements in dissolution rate for raloxifene are reported from liquisolid compacts with cremophor, capmul and transcutol P as non-volatile solvents [31].
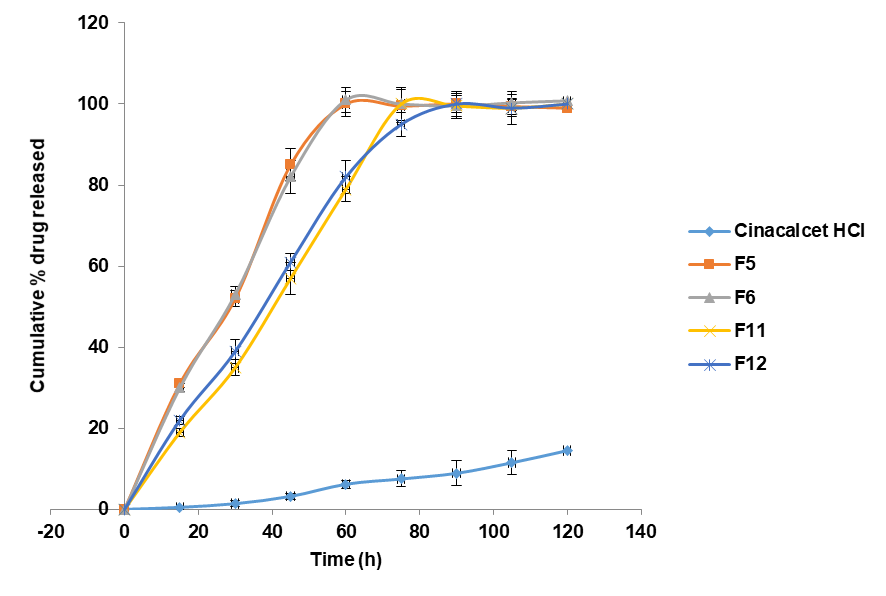
Fig. 2: In vitro dissolution profile for liquisolid tablets, data expresses as mean±SD, n = 6
Table 6: In vitro release kinetic study
| Parameters | CH | F5 | F6 | F11 | F12 |
| Q30 (%) | 1.4±0.02 | 52± 0.5 | 53± 1.2 | 35±1.2 | 39±1.1 |
| DE30 (%) | 1.2± 0.03 | 51.5 ±2.5 | 51.9± 1.3 | 34.2±2.5 | 37.65±3.4 |
| MDT (min) | 44.32± 2.1 | 30.47 ±2.6 | 30.42±3.1 | 27.87±1.6 | 26.65±1.5 |
| Hixson Crowell’s (r2) | 0.834 | 0.967 | 0.998 | 0.998 | 0.998 |
Data expresses as mean±SD, n = 6.
FT-IR study
FT-IR spectroscopic analysis of cinacalcet HCl identified characteristic peaks at 1517 cm⁻¹, indicative of methyl (CH₃) functional groups. Additionally, absorption bands observed at 1338 cm⁻¹, 2909 cm⁻¹, 796 cm⁻¹, and 805 cm⁻¹ corresponded to methylene (CH₂), amine (NH), trifluoromethyl (CF₃), and phenyl (benzene) groups, respectively (fig. 3). The compatibility of cinacalcet HCl with formulation excipients was supported by the presence of analogous spectral features in the liquisolid system.
DSC study
An evident endothermic peak resulting from drug melting is visible in the thermogram of pure cinacalcet HCl. The presence of a strong endothermic peak and a narrow melting range confirms the crystalline form of cinacalcet HCl (fig. 4). The DSC for both tween 80 and labrasol-based liquisolid formulation did not manifest any melting peak, which can be ascribed to the presence of cinacalcet HCl in liquid form i. e. dissolved or molecular state. Similar results are also reported for domperidone [32] and simvastatin [33] i. e. presence of the drug in the molecular state.
P-XRD study
P-XRD diffractogram (fig. 5) for cinacalcet HCl suggests that it is a crystalline drug as it has shown peaks at 2Ꝋ angles of 13, 15, 21, and 25. The diffractograms for powdered samples of both liquisolid tablets (F5 and F12) completely disappeared i. e. no peaks were observed at any of the 2Ꝋ angles. This complete absence of peaks for tween 80 and labrasol-based liquisolid formulations can be imputed to the presence of cinacalcet HCl in solubilized (molecular) form. The results of the P-XRD study corroborated with the results obtained from the DSC study.
Stability study
Liquisolid tablets (F5) did not exhibit any significant change in quality control parameters during 6 mo of stability study as per ICH guidelines (table 7).
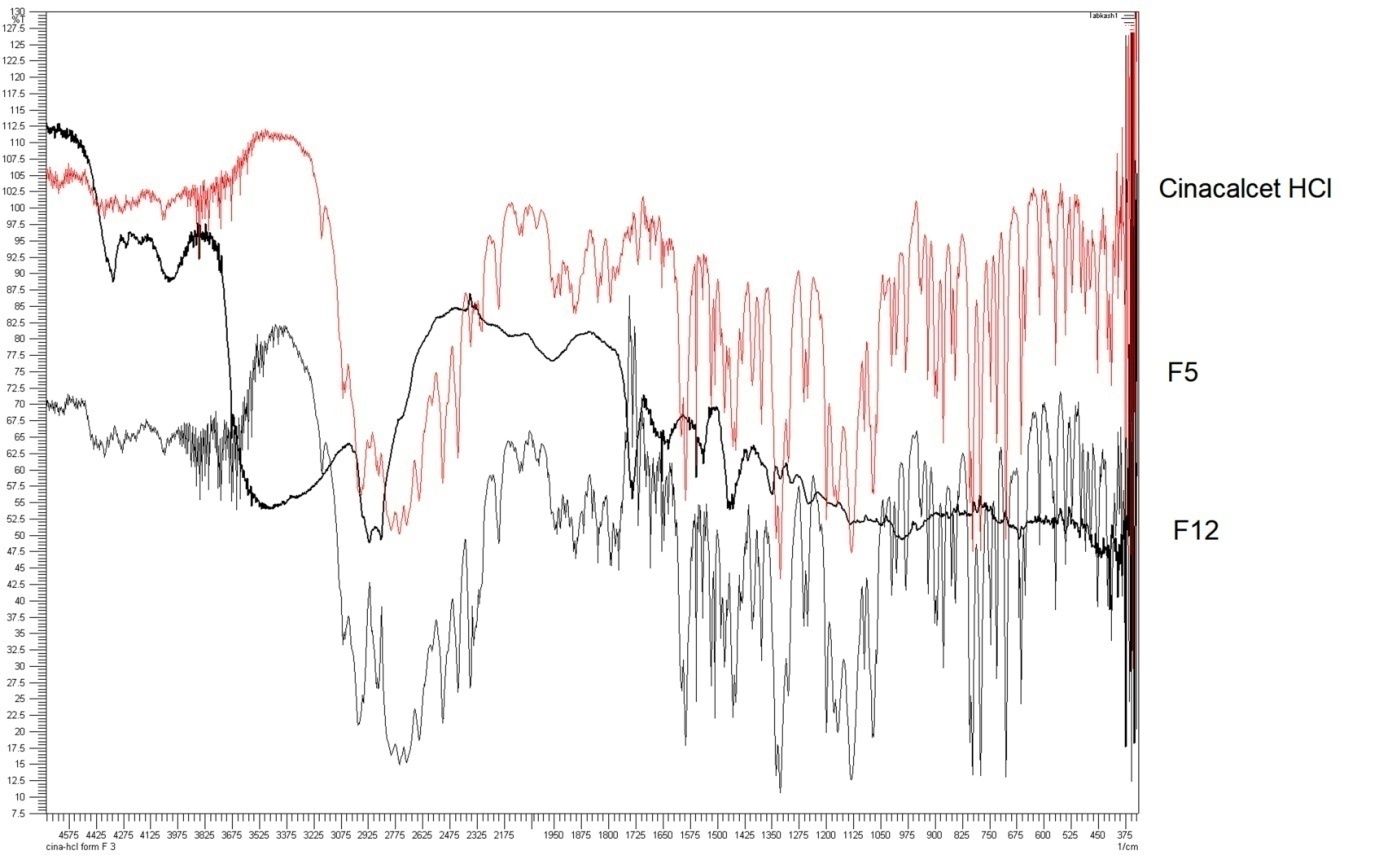
Fig. 3: FT-IR study for cinacalcet HCl, F5 and F12
Pharmacokinetic study
The pharmacokinetic study findings (table 8 and fig. 6) for aqueous suspension of cinacalcet HCl and liquisolid tablet (F5) suggest that tween 80-based liquisolid tablet (F5) exhibited faster dissolution and rapid absorption as ascertained from the decreased Tmax from 6 to 2 h. The intensity of action for the liquisolid tablet (F5) was nearly 3 times more as evidenced by Cmax values. The AUC (area under the curve) values are higher for liquisolid tablets which suggests that oral bioavailability was increased by 2.5 fold [34].
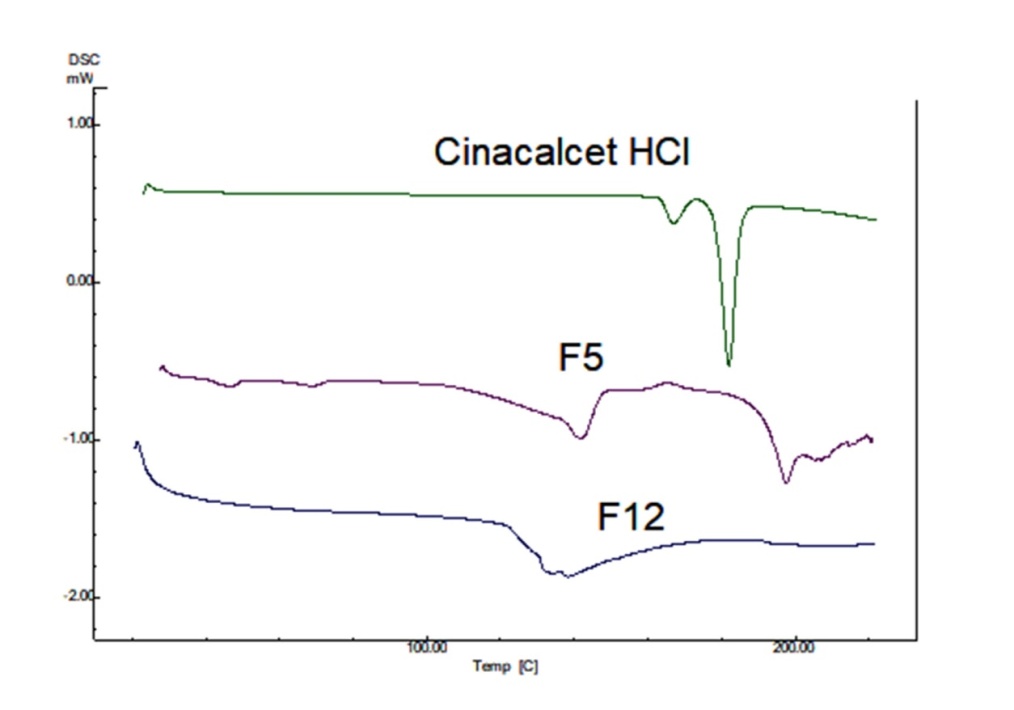
Fig. 4: DSC thermogram for cinacalcet HCl, F5 and F12
Table 7: Stability study of liquisolid tablet (F5)
| Time (mo) | Drug content (%w/w) | Disintegration time (min) | Drug release at 30 min (%) |
| 1 | 96.3±1.7 | 4.1±0.3 | 52.5±0.51 |
| 2 | 96.1±4.85 | 3.8±0.5 | 51.6±0.52 |
| 3 | 95.6±4.82 | 4.1±0.4 | 51. 8±0.56 |
| 6 | 96.2±3.56 | 3.9± 0.5 | 53.9±0.51 |
Data expresses as mean±SD, n = 6.
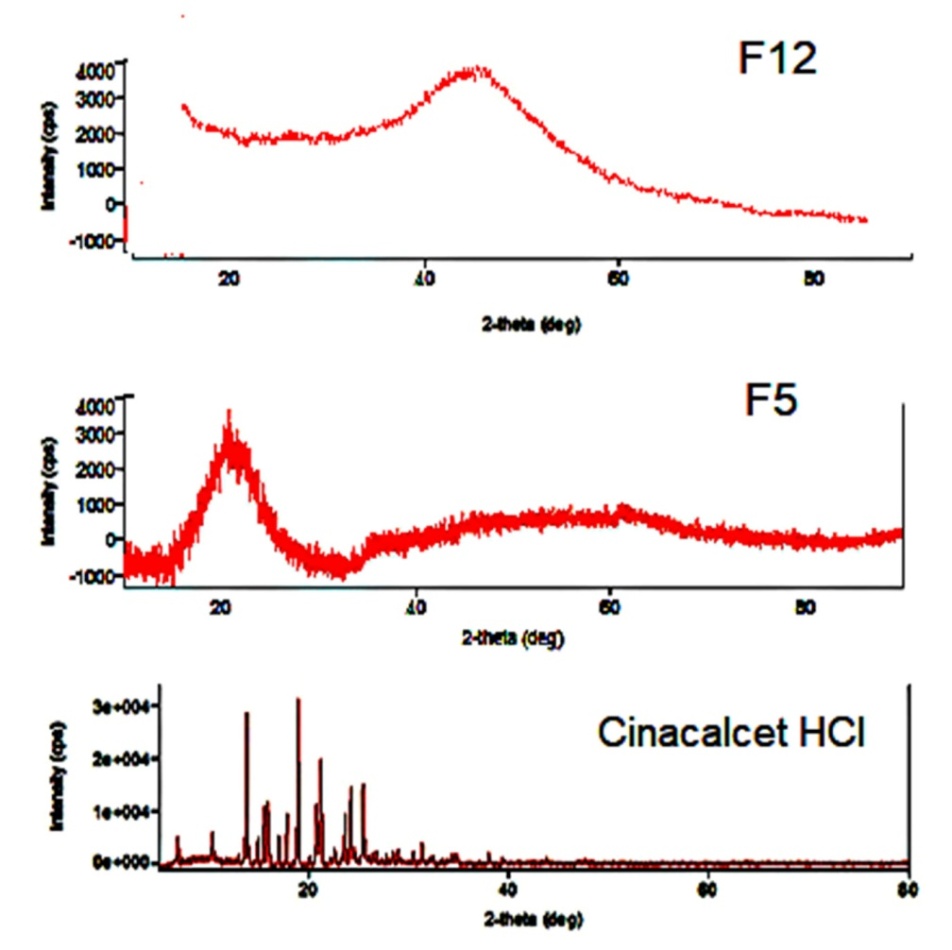
Fig. 5: P-XRD for cinacalcet HCl, F5 and F12
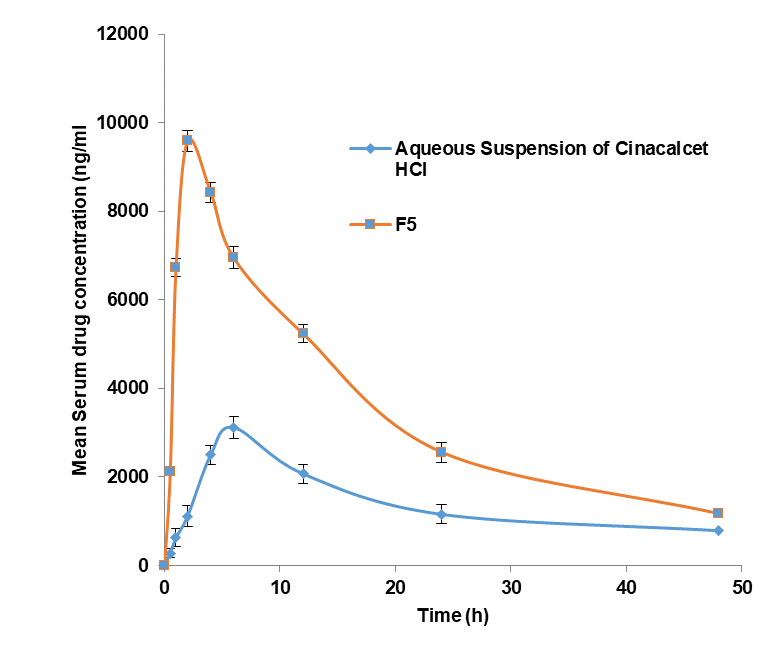
Fig. 6: Serum drug concentration versus time curve for aqueous suspension of CH and F5. Data expresses as mean±SD, n = 6
Table 8: Pharmacokinetic evaluation of liquisolid tablet
| PK parameters | Aqueous suspension of cinacalcet HCl | Liquisolid tablet (F5) |
| Cmax (ng/ml) | 3117.08±36.5 | 9590.7±45.2 |
| Tmax (h) | 6±0.2 | 2.0± 0.1 |
| AUC (ng. h/ml) | 9563.11±89.4 | 21896.86±152.3 |
Data expresses as mean±SD, n = 6.
CONCLUSION
Non-volatile solvents Tween 80 and Labrasol were successfully employed in the creation of liquisolid formulations Notably, the Tween 80-based liquisolid formulation demonstrated significant improvements in solubility and dissolution rate. The porous carrier Neusilin US2, coated with aerosil (R = 20), exhibited remarkable liquid retention capacity. These liquisolid formulations also displayed desirable flowability for tablet processing. Among them, formulation F5 exhibited higher dissolution and disintegration in 4 min. Furthermore, the pharmacokinetic study revealed a 2.5fold enhancement in oral bioavailability for liquisolid formulation F5. Thus, the successful application of the liquisolid technique can enhance both the dissolution rate and oral bioavailability of cinacalcet HCl.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
M. Somesu performed the practical experimental work. Ch. Niranjan Patra guided the candidate while executing the work. Goutam Kumar Jena assisted us in the pharmacokinetic study. Dipti Shree assisted in the interpretation of the DSC and P-XRD study. Sudarsan Biswal contributed to writing the entire manuscript systematically and scientifically.
CONFLICT OF INTERESTS
Declared none
REFERENCES
Hermans A, Milsmann J, LI H, Jede C, Moir A, Hens B. Challenges and strategies for solubility measurements and dissolution method development for amorphous solid dispersion formulations. AAPS J. 2022;25(1):11. doi: 10.1208/s12248-022-00760-8, PMID 36513860.
Savjani KT, Gajjar AK, Savjani JK. Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012 Jul 5;2012:195727. doi: 10.5402/2012/195727, PMID 22830056.
Sruti J, Niranjan Patra C, Swain S, Panigrahi KC, Patro AP, Beg S. Improvement in the dissolution rate and tableting properties of cefuroxime axetil by melt granulated dispersion and surface adsorption. Acta Pharm Sin B. 2013;3(2):113-22. doi: 10.1016/j.apsb.2013.01.001.
Agustina R, Setyaningsih D. Solid dispersion as a potential approach to improve dissolution and bioavailability of curcumin from turmeric (Curcuma longa L.). Int J App Pharm. 2023;15(5):37-47. doi: 10.22159/ijap.2023v15i5.48295.
Paczkowska M, Mizera M, Szymanowska Powalowska D, Lewandowska K, Blaszczak W, Goscianska J. β-Cyclodextrin complexation as an effective drug delivery system for meropenem. Eur J Pharm Biopharm. 2016;99:24-34. doi: 10.1016/j.ejpb.2015.10.013, PMID 26592156.
Sogali BS, BN V, Murthy KV R. Comparative studies with different cyclodextrin derivatives in improving the solubility and dissolution of saquinavir. Asian J Pharm Clin Res. 2018 Jun;11(6)509-16. doi: 10.22159/ajpcr.2018.v11i6.24836.
Wang B, Xiang J, HE B, Tan S, Zhou W. Enhancing the bioavailability of natural extracts for nutritional applications through dry powder inhalers (DPI) spray drying: technological advancements and future directions. Front Nutr. 2023;10:1190912. doi: 10.3389/fnut.2023.1190912, PMID 37476406.
Taldaev A, Pankov DI, Terekhov RP, Zhevlakova AK, Selivanova IA. Modification of the physicochemical properties of active pharmaceutical ingredients via lyophilization. Pharmaceutics. 2023;15(11):2607. doi: 10.3390/pharmaceutics15112607, PMID 38004585.
Rasenack N, Muller BW. Dissolution rate enhancement by in situ micronization of poorly water-soluble drugs. Pharm Res. 2002;19(12):1894-900. doi: 10.1023/a:1021410028371, PMID 12523671.
Shi NQ, Zhang H, Zhang Y, Feng B, LI ZQ, QI XR. Study on the properties of felodipine solid dispersions prepared by different technologies. Beijing DA Xue Xue Bao Yi Xue Ban. 2016;48(6):1067-73. PMID 27987515.
Spireas S. Liquisolid systems and methods of preparing same; 2002.
LU M, Xing H, Jiang J, Chen X, Yang T, Wang D. Liquisolid technique and its applications in pharmaceutics. Asian J Pharm Sci. 2017;12(2):115-23. doi: 10.1016/j.ajps.2016.09.007, PMID 32104320.
Kanojiya PS, Ghodake PN, Wadetwar RN. Design and optimization of liquisolid compact-based vaginal sustained release tablet of antifungal agent for vaginal candidiasis. J Dispers Sci Technol. 2024;45(3):1-16. doi: 10.1080/01932691.2022.2158854.
Panda S, Varaprasad R, Priyanka K, Swain RP. Liquisolid technique: a novel approach for dosage form design. Int J App Pharm. 2017 May;9(3):8. doi: 10.22159/ijap.2017v9i3.18698.
Franceschini N, Joy MS, Kshirsagar A. Cinacalcet HCl: a calcimimetic agent for the management of primary and secondary hyperparathyroidism. Expert Opin Investig Drugs. 2003;12(8):1413-21. doi: 10.1517/13543784.12.8.1413, PMID 12882626.
Padhi D, Harris R. Clinical pharmacokinetic and pharmacodynamic profile of cinacalcet hydrochloride. Clin Pharmacokinet. 2009;48(5):303-11. doi: 10.2165/00003088-200948050-00002, PMID 19566113.
Aleksic I, Glisic T, Cvijic S, Parojcic J. Liquisolid systems: evaluation of the influence of formulation variables on the optimum liquid load. Archives of Pharmacy. 2022;72(1):61-76. doi: 10.5937/arhfarm72-33130.
Sinko PJ. Martins physical pharmacy and pharmaceutical sciences. Lippincott Williams & Wilkins; 2023.
Patra CN, Swain S, Mahanty S, Panigrahi KC. Design and characterization of aceclofenac and paracetamol spherical crystals and their tableting properties. Powder Technol. 2015 Apr;274:446-54. doi: 10.1016/j.powtec.2015.01.053.
Patra ChN, Pandit HK, Singh SP, Devi MV. Applicability and comparative evaluation of wet granulation and direct compression technology to Rauwolfia serpentina root powder: a technical note. AAPS Pharm Sci Tech. 2008;9(1):100-4. doi: 10.1208/s12249-007-9015-7, PMID 18446468.
Lachman L, Lieberman HA, Kanig JL. The theory and practice of industrial pharmacy. Philadelphia: Lea & Febiger; 1976.
Bruschi ML. Strategies to modify the drug release from pharmaceutical systems. Woodhead Publishing; 2015.
Haleem RM, Salem MY, Fatahallah FA, Abdelfattah LE. Quality in the pharmaceutical industry a literature review. Saudi Pharm J. 2015;23(5):463-9. doi: 10.1016/j.jsps.2013.11.004, PMID 26594110.
Routray SB, Patra CN, Swain S, Jena BR. Analytical quality by design-based systematic development and optimization of a sensitive bioanalytical method for estimation cinacalcet HCl in rabbit serum. J Pharm Bioallied Sci. 2021;13(4):360-6. doi: 10.4103/jpbs.jpbs_604_21, PMID 35399796.
Nakaweh A, Al Akayleh F, Al Remawi M, Abdallah Q, Agha AS. Deep eutectic system based liquisolid nanoparticles as drug delivery system of curcumin for in vitro colon cancer cells. J Pharm Innov. 2024;19(2):1-11. doi: 10.1007/s12247-024-09826-w.
Panigrahi KC, Patra CN, Rao ME. Quality by design enabled development of oral self-nanoemulsifying drug delivery system of a novel calcimimetic cinacalcet HCl using a porous carrier: in vitro and in vivo characterisation. AAPS Pharm Sci Tech. 2019;20(5):216. doi: 10.1208/s12249-019-1411-2, PMID 31172322.
Williams HD, Van Speybroeck M, Augustijns P, Porter CJ. Lipid-based formulations solidified via adsorption onto the mesoporous carrier neusilin® US2: effect of drug type and formulation composition on in vitro pharmaceutical performance. J Pharm Sci. 2014;103(6):1734-46. doi: 10.1002/jps.23970, PMID 24740767.
Gumaste SG, Dalrymple DM, Serajuddin AT. Development of solid SEDDS V: compaction and drug release properties of tablets prepared by adsorbing lipid-based formulations onto neusilin® US2. Pharm Res. 2013;30(12):3186-99. doi: 10.1007/s11095-013-1106-4, PMID 23797463.
Kominova P, Kulaviak L, Zamostny PS. Stress-dependent particle interactions of magnesium aluminometasilicates as their performance factor in powder flow and compaction applications. Materials (Basel). 2021;14(4):900. doi: 10.3390/ma14040900, PMID 33672812.
Rao KR, Nagabhushanam MV, Chowdary KP. In vitro dissolution studies on solid dispersions of mefenamic acid. Indian J Pharm Sci. 2011;73(2):243-7. doi: 10.4103/0250-474x.91575, PMID 22303074.
Komala DR, Janga KY, Jukanti R, Bandari S, Vijayagopal M. Competence of raloxifene hydrochloride loaded liquisolid compacts for improved dissolution and intestinal permeation. J Drug Deliv Sci Technol. 2015 Dec;30:232-41. doi: 10.1016/j.jddst.2015.10.020.
Bhatia M, Rani G, Bahmani K, Devi S. Formulation and optimization: liquisolid of domperidone for solubility enhancement. Acta Pharm Sci. 2024;62(3). doi: 10.23893/1307-2080.APS6234.
Dindigala AK, Reddy CS, Makineni A. Enhancement of solubility and dissolution rate of simvastatin tablets by liquisolid compact approach. J Drug Delivery Ther. 2024;14(8):64-72. doi: 10.22270/jddt.v14i8.6733.
Patra CN, Mishra A, Jena GK, Panigrahi KC, Sruti J, Ghose D. QBD enabled formulation development of nanoemulsion of nimodipine for improved biopharmaceutical performance. J Pharm Innov. 2023;18(3):1279-97. doi: 10.1007/s12247-023-09714-9.