
Int J App Pharm, Vol 17, Issue 2, 2025, 359-375Original Article
IN SILICO, DEVELOPMENT AND CHARACTERIZATION OF CILNIDIPINE ANALOGUES FOR ENHANCED SOLUBILITY AND THERAPEUTIC POTENTIAL IN THE TREATMENT OF ANGINA PECTORIS
PAVANKUMAR KROSURI1, MOTHILAL MOHAN2*
1SRM College of Pharmacy, SRM Institute of Science and Technology, Kattankulathur, Chengalpattu-603203, Tamilnadu, India. 2Department of Pharmaceutics, SRM College of Pharmacy, SRM IST, Kattankulathur, Chengalpattu-603203, Tamilnadu, India
*Corresponding author: Mothilal Mohan; *Email: mothipharma78@gmail.com
Received: 06 Sep 2024, Revised and Accepted: 14 Dec 2024
ABSTRACT
Objective: Angina pectoris remains a significant clinical challenge due to the limitations of current therapies, such as inadequate efficacy and undesirable side effects. This study aims to address these issues by developing a novel treatment approach based on Cilnidipine analogues and nanotechnology, focusing on the lead compound Cilnidipine Analogue (CC5; ZINC101069658).
Methods: CC5 was identified as the most promising candidate due to its optimal balance of lipophilicity, solubility, absorption, and synthetic feasibility. A bioinformatics-driven approach uncovered 60 potential target hub genes related to angina pectoris, with Protein-Protein Interaction (PPI) analysis highlighting Estimated Glomerular Filtration Rate (EGFR) as a key target. Molecular docking and Molecular Dynamics simulations confirmed the stability and strong binding affinity of CC5 with the EGFR-associated protein (5wb7). SWISSADME analysis revealed moderate lipophilicity, poor water solubility, and low gastrointestinal absorption, while HOMO-LUMO studies suggested enhanced chemical stability. The Taguchi design of experiments indicated that stirring speed was critical for nanoparticle size, and stabilizer concentration significantly impacted Encapsulation Efficiency (EE) and zeta potential.
Results: Molecular docking studies showed a strong binding affinity of-8.6 kcal/mol with EGFR, while pharmacokinetic evaluations indicated favourable absorption and moderate lipophilicity, supporting CC5’s potential as an optimized therapeutic agent for angina pectoris. CC5 nanoparticles exhibited a 2.63-fold increase in solubility compared to the parent compound. Fourier Transform Infrared Spectroscopy, Transmission Electron Microscopy, and X-ray Diffraction characterization confirmed the successful nanoparticle formulation. In vitro dissolution studies demonstrated superior drug release from CC5-loaded nanoparticulate oral disintegrating tablets, with the CAF9 (CC5 Formulation 9) showing rapid onset of action and a significantly improved release profile (98.89±1.10% at 30 min) compared to controlled (80.58%) and marketed preparations (18.85%).
Conclusion: The study demonstrates the therapeutic potential of ZINC101069658, a CC5, through its enhanced solubility and reduced Crystallinity. The lead compound was made into Nanoparticles using Pluronic F 188 as carrier. These nanoparticles were further formulated to oral disintegrating tablets for rapid drug release good stability compared to conventional tablets. These findings suggest that ZINC101069658 could be a promising candidate for the treatment of angina pectoris.
Keywords: Cilnidipine analogues, Drug discovery, Toxicity, Nanoparticles, Taguchi design
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i2.52603 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Angina Pectoris (AP) is a prevalent clinical syndrome caused by myocardial ischemia, often due to epicardial coronary artery stenosis, microvascular dysfunction, or coronary vessel constriction, and can progress to Coronary Artery Disease (CAD) [1]. Globally, AP significantly impacts healthcare, contributing to increased morbidity and mortality. Standard therapeutic interventions, including nitrates, beta-blockers, calcium channel blockers, aspirin, and Angiotensin-Converting Enzyme (ACE) inhibitors, aim to alleviate ischemic symptoms and improve patient outcomes. However, the efficacy of current treatments is limited, and many patients experience suboptimal therapeutic responses or undesirable side effects, necessitating the development of novel therapies with improved efficacy and safety profiles [2].
Among calcium channel blockers, Cilnidipine, a third-generation dihydropyridine, stands out due to its unique dual L/N-type calcium channel antagonism, which offers advantages in managing hypertension and AP compared to other calcium channel blockers that only target L-type channels [18, 19]. Despite its clinical effectiveness, Cilnidipine's therapeutic potential is hindered by poor aqueous solubility, which limits its bioavailability and overall clinical efficacy [20]. Addressing this limitation could significantly enhance treatment outcomes for patients with angina pectoris.
The drug discovery and development process is notoriously complex, with high costs, long development times, and significant risks. Even after successful target identification and hit validation, candidates often face challenges related to pharmacokinetics, efficacy, and toxicity during clinical trials [3]. The journey from synthesis to the first human trial can span over 2.6 y, costing approximately USD $430 million, and clinical testing through regulatory submission adds another 6 to 7 y [4]. Given increasing regulatory scrutiny from bodies like the Food and Drug Administration (FDA), the failure rates in clinical trials have risen, further escalating Research and Development (R and D) costs [5].
Advancements in Computer-Aided Drug Design (CADD) have revolutionized the traditional drug discovery process, enabling more efficient target identification, hit discovery, and lead optimization. CADD integrates disease pathway identification, target protein prediction, and ligand discovery through database searches such as ZINC, coupled with in silico preclinical assessments of pharmacokinetics and toxicity using platforms like Swiss ADME and PROTOX 3.0 [7-9]. These methodologies significantly shorten the timeline for lead optimization and reduce costs compared to traditional approaches, which can extend over 15-16 y [6].
A key challenge in drug development is poor solubility and bioavailability, both of which are crucial for the therapeutic efficacy of a drug. Drug absorption is largely dependent on dissolution in biological fluids, and approximately 40% of chemical entities suffer from poor aqueous solubility, resulting in high dosage requirements, variable absorption, and delayed therapeutic effects [10, 11]. To overcome these limitations, various strategies-such as co-crystallization, co-solvent systems, solid dispersions, and nanosizing-have been employed to improve solubility and bioavailability [12, 13].
Nanotechnology has emerged as a powerful tool in drug delivery, offering enhanced chemical, biological, and physical properties that improve drug dissolution, targeting, and therapeutic outcomes [14, 15]. Nanoparticle-based drug delivery systems, characterized by optimized particle size and surface properties, ensure more precise and effective delivery of therapeutic agents to target tissues, addressing the solubility and bioavailability challenges that hinder conventional formulations [16, 17].
In this study, we hypothesize that Cilnidipine analogues, specifically CC5 (ZINC101069658), with modified physicochemical properties, will offer enhanced therapeutic efficacy for angina pectoris. By leveraging CADD to identify and optimize the lead compound, and applying nanotechnological approaches to improve solubility and bioavailability, this study aims to develop a more effective treatment option for angina pectoris. Additionally, EGFR was identified as a key target for CC5 through bioinformatics and Protein-Protein Interaction (PPI) analysis, providing further rationale for its potential efficacy in the treatment of angina pectoris.
MATERIALS AND METHODS
Materials
Lead Compound was obtained from AK scientific, Transcutol HP, Pluronic F188, Kyron T 314 was obtained from Research labs-Fine chem Industries, Raw Banana Powder was obtained from vital Herbs, Delhi, methyl carboxy cellulose, Magnesium stearate, Talc, NaoH were obtained from SD fine chemicals, Mumbai.
Methods
Computational studies were conducted using a combination of the ZINC database [21], PyRx, and Discovery Studio Visualizer v21.1.0 [22, 23]. Initial ligand identification was performed using the ZINC database, a comprehensive repository of commercially available compounds. For molecular docking simulations, PyRx was employed to dock these ligands with the target protein, generating various binding poses and predicting binding affinities. The docking results were analysed and visualized with Discovery Studio Visualizer v21.1.0, providing insights into binding interactions and the stability of ligand-protein complexes. Additional online resources, including PASS, Protox, OSIRIS Property Explorer, SWISS ADME, and pkcsm, were used to predict pharmacokinetics, toxicity, and drug-like properties of the compounds [24, 25]. The Protein Data Bank was also consulted for structural data on the target proteins. This integrated approach facilitated a comprehensive evaluation of potential drug candidates, optimizing the selection process for further development.
Identification of lead compound
The process of identifying the lead compound from Cilnidipine derivative involved several key steps. Initially, data on Cilnidipine was gathered from the PubChem database to understand its biomolecular activities. Compounds for virtual screening were sourced from the ZINC database. Active compound target prediction was performed using the Swiss Target Prediction tool, which helped identify potential biological targets. Subsequently, drug-like properties and absorption parameters of the compounds were assessed using the Swiss ADME platform, focusing on gastrointestinal absorption and adherence to drug-likeness criteria.
Selection of target hub genes via network pharmacology
Data acquisition
To identify genes implicated in angina pectoris, we systematically mined three well-established databases: Online Mendelian Inheritance in Man (OMIM), Gene Cards, and STITCH (Search Tool for Interactions of Chemicals). Gene sets associated with AP were extracted from each database to compile a comprehensive pool of candidate genes for subsequent analysis [26, 27].
Venn diagram analysis
The gene sets obtained from OMIM, Gene Cards, and STITCH were subjected to comparative analysis through Venn diagram construction. This approach facilitated the identification of overlapping genes across the three datasets. The intersecting genes were considered as candidate target hub genes potentially implicated in angina pectoris.
PPI network analysis
PPI network construction
The candidate hub genes identified from the Venn diagram analysis were further investigated to elucidate their PPI using the STRING database (version 12.0). A PPI network was constructed to delineate the interaction landscape of the proteins encoded by these genes. An interaction confidence threshold of ≥0.7 was employed to ensure the inclusion of high-confidence interactions within the network [28].
PPI network visualization
The PPI network was visualized using Cytoscape software (version 3.10.3), enabling the identification of key interaction hubs within the network. In this visualization, nodes represent proteins, and edges denote the interactions between them [28].
Molecular docking and molecular dynamics
Molecular docking
Molecular docking studies were conducted to predict the binding affinity and orientation of the lead compound (CC5, ZINC101069658) with Estimated Glomerular Filtration Rate (EGFR) (PDB ID: 5WB7). The protein structure was prepared by adding missing hydrogen atoms, removing water molecules, and optimizing protonation states using Auto Dock Tools 1.5.6. The grid box for docking was defined to encompass the active site of the target protein with the following parameters: grid dimensions of 60 × 60 × 60 Å centered at X = 12.45, Y =-0.23, Z = 36.18, ensuring coverage of the binding pocket. Exhaustiveness was set to 8 to ensure sufficient sampling of ligand conformations. Ligand preparation was carried out using LigPrep in Schrödinger with the OPLS3e force field, optimizing bond lengths and minimizing the ligand's energy. Flexible ligand docking was performed using the SP (Standard-Precise) scoring function in Auto Dock Vina 1.5.6, which predicted the binding energies of the ligand-receptor complexes.
Visualization and analysis of the docking poses were performed using Discovery Studio Visualizer v21.1.0, allowing detailed examination of key interactions such as hydrogen bonding, hydrophobic contacts, and π-π stacking interactions between CC5 and the EGFR binding site [29].
Molecular dynamics simulations
Molecular dynamics simulations were performed using the Desmond module within the Schrödinger suite v2021-3 to evaluate the stability of the CC5-EGFR complex over time. The OPLS3e force field was used to model molecular interactions. The system was solvated in a cubic water box with a 10 Å buffer distance using the TIP3P water model. Sodium and chloride ions were added to neutralize the system, with a final salt concentration of 0.15 M. The molecular dynamics simulation was run at a constant temperature of 300 K and a pressure of 1 atm using the NPT ensemble. The simulation was conducted for a duration of 100 ns with a time step of 2 fs, and periodic boundary conditions were applied. The results, including Root mean Square Deviation (RMSD) and Root mean Square Fluctuation (RMSF) analyses, were computed to evaluate the stability and conformational flexibility of the ligand-receptor complex [29].
Validation of computational models
Docking validation was performed by re-docking a known ligand, quercetin, into the EGFR binding site, which achieved an RMSD of less than 2.0 Å compared to the original co-crystallized pose, confirming the accuracy of the docking protocol. For molecular dynamics simulations, experimental data from literature on EGFR-inhibitor binding stability were used to corroborate the molecular dynamics results, ensuring the reliability of the simulated dynamics of the ligand-receptor complex.
Estimation of pharmacokinetics, drug-likeness properties and toxicity
Pharmacokinetic properties, drug-likeness, and toxicity profiles of the compounds were evaluated to identify promising drug candidates and minimize experimental testing. Drug-likeness was estimated using Lipinski's Rule of Five via the Swiss ADME platform. Compounds were analyzed by entering their Simplified Molecular Input Line Entry System (SMILES) format into the Swiss ADME server, which generated their ADME profiles and drug-likeness parameters. Toxicity assessments were performed using Protox and Osiris online tools [30].
HOMO and LUMO study
To analyze the electronic properties and stability of the compound, we conducted a Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) study, focusing on the HOMO LUMO energies. Initially, the compound was modeled and optimized using molecular modeling software to achieve stable conformations. Quantum mechanical calculations, using methods such as Density Functional Theory (DFT), were then performed to obtain the HOMO and LUMO energy levels [31].
The HOMO-LUMO gap was calculated as the difference between these energy levels, providing insights into the compound's stability and reactivity. A smaller gap generally indicates greater stability and lower reactivity, while a larger gap suggests higher reactivity. Molecular orbitals were visualized using tools like Avogadro or ChemDraw to examine the electron density distribution and active sites.
Finally, the HOMO-LUMO gap was compared with values from similar compounds to assess relative stability and reactivity. These results were contextualized within the broader scope of chemical interactions and potential applications, guiding further research and development efforts.
Determination of saturated solubility
Saturated solubility of both the parent drug and the lead compound was assessed by dissolving an excess amount of each powder in 5 ml of phosphate buffer (pH 6.8) within separate vials. The vials were placed in a thermostatically controlled shaking water bath (Precision Scientific Inc., Chicago, IL, USA) at 37 °C and agitated for 48 h to ensure equilibrium. Following agitation, samples were filtered through a 0.1 μm membrane filter (Whatman Inc., Clifton, NJ, USA). The filtrate was analysed using a spectrophotometer at the predetermined wavelength of 240 nm, employing disposable micro cuvettes. The process was repeated in triplicate for each compound, and the solubility was reported as mean values±standard deviation [32].
Differential scanning calorimetry (DSC)
DSC analysis was performed with two milligrams of dried nanoparticle samples using a temperature range of 50 °C to 450 °C, a nitrogen flow rate of 20 ml/min, and a heating rate of 20 °C/min to assess thermal properties [33].
Fourier transform infrared spectroscopy (FTIR)
FTIR analysis was conducted using a Jasco FT/IR 4700 spectrometer. Samples, including plain Cilnidipine, Lead compound-loaded nanoparticles, and empty nanoparticles, were placed on an ATR crystal. Spectra were recorded to investigate the presence of Cilnidipine derivatives in the nanoparticles [34].
X-ray diffraction (XRD)
XRD was utilized to determine the crystallinity and structural characteristics of the samples, providing insights into their crystal structure [35].
Development of lead compound-loaded nanoparticles
The antisolvent precipitation method was selected for the synthesis of lead compound-loaded nanoparticles due to its suitability for enhancing the solubility of poorly water-soluble drugs. This method offers several advantages over alternative nanoparticle formulation techniques such as milling or spray drying. Antisolvent precipitation allows for better control over particle size, prevents degradation due to heat (as opposed to spray drying), and avoids the mechanical stress associated with milling, which can affect the drug’s stability and efficacy. Moreover, this technique is cost-effective and scalable, making it suitable for industrial applications.
In this process, the drug is first dissolved in a solvent (Transcutol HP), and then rapidly introduced into a miscible antisolvent (water) containing a stabilizer/Polymer such as Pluronic F 188. The rapid mixing induces supersaturation, promoting the nucleation and formation of fine drug particles. The process requires fine-tuning to optimize key parameters such as nucleation and crystal growth rates, as well as particle stabilization, to ensure uniformity and stability of the nanoparticles.
The optimization of nanoparticle synthesis was performed using a Taguchi design approach. Variables such as stirring speed, stabilizer concentration, and solvent-to-antisolvent ratio were systematically varied to identify the optimal conditions for minimizing particle size and maximizing entrapment efficiency (EE). For instance, higher stirring speeds were found to enhance homogenization, reducing particle size, while the concentration of stabilizers influenced the zeta potential and stability of the nanoparticles. The Taguchi design methodology helped in identifying the most significant factors and their optimal levels for achieving the desired nanoparticle characteristics [36].
Design of experiments
Table 1 outlines the Taguchi design variables employed in the preparation of Lead Compound-loaded nanoparticles. The independent variables and their levels are as follows: the antisolvent-to-solvent ratio (Factor A) is set at 2.00, 6.00, and 10.00 for low, medium, and high levels, respectively; the stabilizer concentration (Factor B) ranges from 1.00% to 5.00%; and the stirring speed (Factor C) is varied from 2000 RPM to 10000 RPM. The dependent variables assessed are particle size (R1), EE (R2), and zeta potential (R3). Experimental runs were created and analysed with Minitab 19.
Table 1: Independent variables with their levels and response variables of lead compound loaded nanoparticle preparation using taguchi design
| Independent factors | Name | Units | Level 1 (-1) | Level 2 (0) | Level 3 (+1) | Response | Units |
| A | Stabilizer Concentration | % | 1 | 3 | 5 | Particle size | nm |
| B | Stirring Speed | RPM | 2000 | 6000 | 10000 | EE | % |
| C | Temperature | °C | 25 | 40 | 55 | Zeta potential | mV |
| D | Solvent: Antisolvent Ratio | v/v | 1:5 | 1:15 | 1:25 | Drug Release | % |
Characterization of nanoparticles
Characterization of Lead Compound-loaded nanoparticles involved several key measurements. The size and zeta potential of the nanoparticles were assessed using a Malvern Zetasizer Nano ZS (Malvern Instruments, UK). This instrument equipped with a 173 °C backscattered light detector and the mean particle size (nm) using Dynamic Light Scattering (DLS). Zeta potential values (mV) were determined via Laser Doppler anemometry. All measurements were conducted in triplicate at 25 °C. DSC and FTIR and Transmission Electron Microscopy (TEM) studies were conducted to characterize the prepared nanoparticles [37].
EE
The % EE of Lead Compound-loaded nanoparticles was assessed using an indirect method. After centrifugation at 40 °C and 6,000 rpm for 30 min, the supernatant was analyzed with a UV spectrophotometer (Jasco UV-2400, Japan) at 240 nm to quantify the free drug. The % EE was calculated by comparing the total drug used in nanoparticle preparation to the drug concentration in the supernatant, using a standard curve for reference. Three measurements of the % EE were performed [38].
Cilnidipine analogue loaded nanoparticulate oral disintegrating tablets.
The lead compound-loaded nanoparticles were further formulated into Oral Disintegrating Tablets (ODTs). As outlined in table 2, the formulation components were accurately weighed and thoroughly blended to ensure homogeneous drug distribution. Kyron T 314 and Raw Banana Powder were taken as Superdisintegrants and Lubricants such as magnesium stearate and talc were carefully incorporated into the mixture to enhance tablet compressibility and prevent sticking during manufacturing. The blend was compressed into tablets using a 16-station Cadmach rotary compression machine fitted with 8 mm biconcave punches, producing tablets with consistent hardness and uniform weight distribution.
Quality control tests were conducted to ensure the ODTs met pharmacopeial specifications for hardness, weight variation, and disintegration time. Control experiments compared the nanoparticle-based formulations to a marketed bulk drug formulation, revealing that the Cilnidipine Analogue Loaded nanoparticulate ODTs exhibited a significantly faster dissolution rate and enhanced bioavailability compared to the bulk drug. This improvement is attributed to the reduced particle size and increased surface area of the nanoparticle formulation, which facilitated better drug solubilization and absorption. The enhanced performance of the nanoparticle formulation offers promising potential for improving therapeutic outcomes in clinical settings. The tablets were stored in a moisture-controlled environment to preserve their integrity and efficacy [39].
Table 2: Formulation table for cilnidipine analogue loaded nanoparticulate oral disintegrating tablets
| Formulation/Ingredients (mg) | CAF1 | CAF2 | CAF3 | CAF4 | CAF5 | CAF6 | CAF7 | CAF8 | CAF9 |
| Drug loaded nanoparticles equivalent to 10 mg of cilnidipine analogue | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 | 30 |
| Kyron T 314 | 5 | 10 | 15 | - | - | - | - | - | - |
| Raw banana powder | - | - | - | 5 | 10 | 15 | - | - | - |
| Kyron T 314+Raw banana powder | - | - | - | - | - | - | 5(2.5+2.5) | 10(5+5) | 15(7.5+7.5) |
| Microcrystalline cellulose (MCC) | 161 | 156 | 151 | 161 | 156 | 151 | 161 | 156 | 151 |
| Magnesium stearate | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Talc | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Total(mg) | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 |
In vitro dissolution study
An in vitro dissolution study was performed using 900 ml of pH 6.8 phosphate buffer maintained at 37±0.50 °C with a stirring rate of 50 rpm, utilizing a USP Type II (paddle) dissolution apparatus. At predetermined intervals (0, 5, 10, 15, 20, 25, and 30 min), 5 ml aliquots of the dissolution medium were withdrawn and replaced with an equal volume of fresh buffer to maintain the sink conditions. The drug content was determined by measuring the absorbance of the samples at 240 nm following filtration. The cumulative percentage of drug released was calculated to represent the drug concentration over time [40].
Stability studies
The stability of Lead Compound-loaded nanosized particulate oral disintegrating tablets was assessed over a three-month period. The tablets were stored in tightly sealed, autoclavable transparent glass vials under 25 °C±2 °C/60+5%RH in accordance with ICH guidelines Q1A (R2). At the end of each month, samples were analyzed for particle size (nm) and % EE of the Lead Compound. Additionally, the pH, viscosity, and drug release profile of the Lead Compound-loaded nanoparticles were evaluated [41].
RESULTS
Identification of lead compound
The analysis of Cilnidipine and its derivatives, as presented in the table 3, reveals several key insights into their pharmacokinetic and pharmacodynamic properties. All compounds, including the parent drug Cilnidipine (CC1), share a consistent molecular weight of 492.58 g/mol, indicating that the derivatives involve minor structural changes rather than significant alterations to the molecular framework.
The log P values for these compounds range from 4.2758 (CC1-CC4 and CC7-CC8) to 5.1161 (CC5-CC6). Log P is a crucial factor in determining a compound’s lipophilicity, which influences its ability to permeate cell membranes. CC5 and CC6 exhibit higher log P values, suggesting increased lipophilicity, which could enhance membrane permeability.
Table 3: Key observations of cilnidipine and its derivatives based on zinc database, swiss ADME, pkcsm
| Compound code | Zinc id | Mol. wt. (gm/mol) | Log p | Water solubility (log mol/l) | Intestinal absorption (%) | Synthetic accessibility | Predicted toxicity LD50 (Protox 3.0) |
| CC1 | ZINC19632657 (Cilnidipine) |
492.58 | 4.2758 | -6.671 | 91.359 | 4.76 | 2968 mg/kg):Class 5 |
| CC2 | ZINC19632663 Cilnidipine) | 492.58 | 4.2758 | -6.671 | 91.359 | 4.76 | 2968 mg/kg):Class 5 |
| CC3 | ZINC1903853580 | 492.58 | 4.2758 | -6.671 | 91.359 | 4.76 | 2968 mg/kg):Class 5 |
| CC4 | ZINC1903853577 | 492.58 | 4.2758 | -6.671 | 91.359 | 4.76 | 2968 mg/kg):Class 5 |
| CC5 | ZINC101069658 | 492.58 | 5.1161 | -6.877 | 95.048 | 5.05 | 2968 mg/kg):Class 5 |
| CC6 | ZINC101069663 | 492.58 | 5.1161 | -6.877 | 95.048 | 5.05 | 2968 mg/kg):Class 5 |
| CC7 | ZINC31261357 | 492.58 | 4.2758 | -6.48 | 88.12 | 4.76 | 2968 mg/kg):Class 5 |
| CC8 | ZINC31261360 | 492.58 | 4.2758 | -6.48 | 88.12 | 4.76 | 2968 mg/kg):Class 5 |
Water solubility is another critical factor that affects drug absorption and bioavailability. The negative values observed for water solubility across all compounds (ranging from 6.671 to 6.877). Among all compounds CC5 and CC6 have better water Solubility.
Despite these solubility challenges, the intestinal absorption percentages for all compounds are relatively high, ranging from 95.048% (CC5 and CC6) to 88.12% (CC7 and CC8). This suggests that the compounds are well-absorbed in the intestines, likely due to their lipophilicity. However, it is important to note that high absorption efficiency may not fully translate to bioavailability if solubility remains a limiting factor.
The synthetic accessibility scores for the compounds range from 4.76 to 5.05, indicating that their synthesis is moderately challenging. CC5 and CC6, with slightly higher synthetic accessibility scores, may be more complex to synthesize, possibly due to their increased lipophilicity or structural modifications. Lastly, the predicted toxicity (LD50) for all compounds is consistent, placing them in toxicity class 5 with an LD50 of approximately 2968 mg/kg. This classification suggests a relatively low acute toxicity profile, which is favorable for further development.
Considering the balance between these various properties, CC5 (ZINC101069658) and CC6 (ZINC101069663) emerge as the most promising candidates for further development. Both compounds have a moderate log P value (5.11). They also exhibit slightly better water solubility (-6.877) compared to other compounds, coupled with high intestinal absorption (95.08%). Their synthetic accessibility remains within a manageable range, and their predicted toxicity is in line with the other derivatives. Therefore, CC5 (ZINC101069658) is identified as the lead compound due to its optimal balance of lipophilicity, solubility, absorption, and synthetic feasibility, making it a strong candidate for further preclinical and clinical evaluation in the treatment of angina pectoris.
Identification of target hub genes
Venn diagram analysis
The comparative analysis of gene sets from the OMIM, Gene Cards, Swiss target prediction and STITCH databases identified 48 genes associated with APin OMIM, 4592 genes in Gene Cards, 100 in Swiss target prediction and 1 gene in STITCH. The Venn diagram presented in fig. 1 revealed an intersection of 60 genes common across all three databases. These 60 overlapping genes were identified as potential target hub genes for further analysis.
STRING database analysis and PPI network construction
The 60 candidate hub genes were analyzed for protein-protein interactions using the STRING database. A PPI network was constructed, and it was displayed in fig. 2, comprising 68 nodes (proteins) and 186 edges (interactions), with an interaction confidence score threshold set at ≥0.7. The network analysis highlighted several highly interconnected nodes indicative of potential hub genes within the angina pectoris-related PPI network.
Identification and prioritization of top hub genes
Using the CytoHubba plugin in Cytoscape, the top 10 hub genes within the PPI network were ranked based on their degree of connectivity and centrality as disclosed in fig. 3. Among the top 10 target genes identified for angina pectoris, the SRC gene ranked first. However, its associated protein was not selected for molecular docking studies due to SRC being a non-receptor tyrosine kinase, which typically presents challenges for docking analyses. The next top-ranking gene was EGFR, and its associated protein, identified as 5wb7, was selected for molecular docking studies. This selection was made based on the receptor status of EGFR, which makes it a suitable candidate for docking and subsequent drug-target interaction analysis.
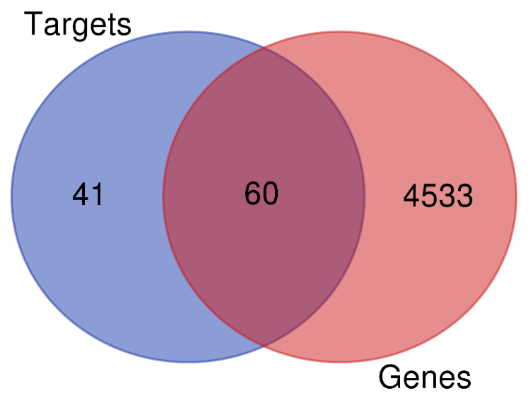
Fig. 1: Venn diagram for identification of target genes for angina pectoris
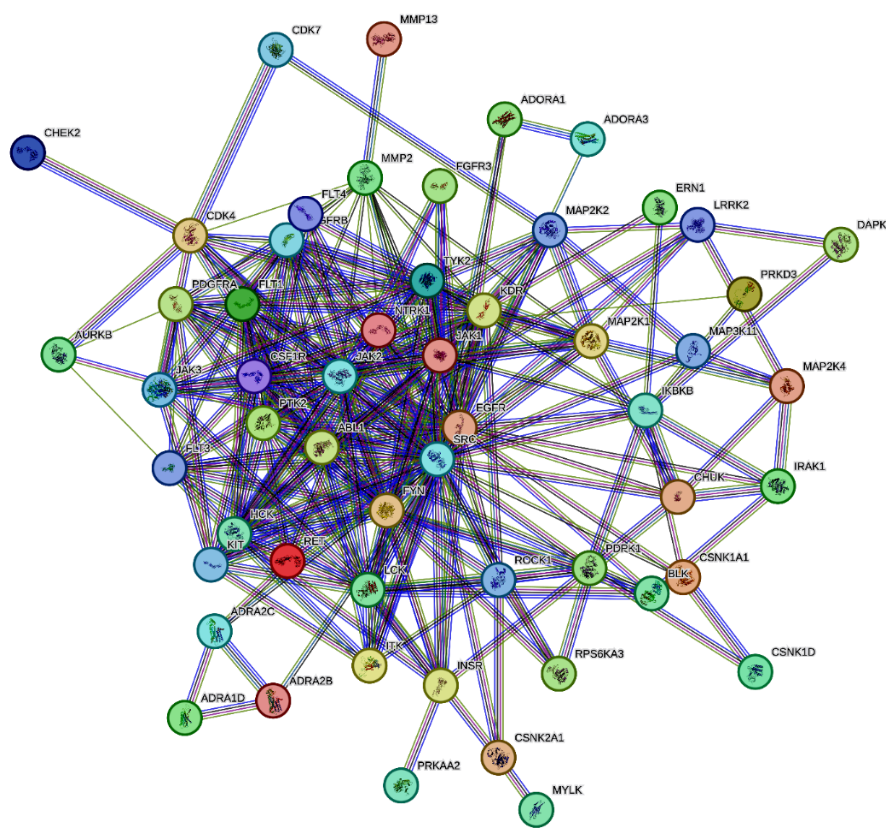
Fig. 2: Protein-protein interactions by string database
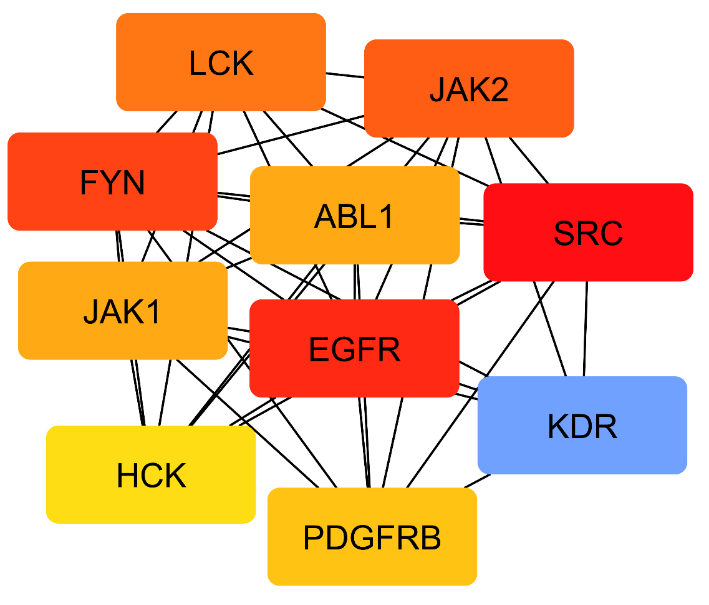
Fig. 3: Top 10 target genes based on cytoscape tool
Molecular docking
The in silico docking study of Cilnidipine derivatives (CC1-CC8) with the EGFR-associated protein 5wb7 revealed various binding affinities and interaction profiles, as summarized in table 4. The binding energy differences among the derivatives correlate with their in vitro efficacy, highlighting their potential as effective inhibitors.
Table 4: Summary of molecular docking results of cilnidipine and its derivatives
| Compounds | Binding affinity score (kcal/mol) | Interacting residues | Type of interactions | Distance |
| CC1 (Parent compound) | -7.4 | GLY C: 264, ASN C: 86 ARG C: 285,ALA C: 286 |
Conventional H-Bond Pi-Alkyl |
2.02,2.53 4.19,4.46 |
| CC2 (Parent compound) | -7.4 | ASP D: 344, THR H: 37 SER D: 11, GLY D: 317, HIS H: 43 ASP D: 323 VAL H: 39 HIS H: 43 |
Conventional H-Bond Carbon H-Bond Pi-Anion Pi-Sigma Pi-Pi T-shaped |
2.18,3.04 2.45,2.75,2.83 3.79 2.82 5.48 |
| CC3 | -8.1 | VAL H: 39, HIS H: 43, HIS D: 409 GLY H: 38, ASN D: 12, GLN D: 408 VAL H: 39 |
Conventional H-Bond Carbon H-Bond Pi-Alkyl |
2.19,2.51,2.95 2.00,2.55,3.07 4.65 |
| CC4 | -6.8 | HIS F: 43, GLN B: 8, HIS B: 409, SER B: 11 GLN B: 408 HOH B: 723 |
Conventional H-Bond Carbon H-Bond Unfavorable |
1.95,2.03,2.05, 2.06 2.50, 1.47 |
| CC5 | -8.6 | THR G: 37, GLY G: 38, HIS G: 43, GLN C: 408 HIS C: 346 LYS C: 13, LEU C: 14,VAL G: 39 |
Conventional H-Bond Pi-Sigma Pi-Alkyl |
2.02,2.32,2.70,293 3.61 4.66,4.80,5.39 |
| CC6 | -7.5 | HIS G: 43, SER C: 324 HIS C: 409 GLU G: 42 PHE C: 380 |
Conventional H-Bond Carbon H-Bond Pi-Anion Pi-Pi T-shaped |
2.68,2.85 2.73 4.81 5.55 |
| CC7 | -7.0 | HIS D: 409, THR H: 37 THR D: 10,SER D: 11 |
Conventional H-Bond Carbon H-Bond |
2.11,2.39 2.49,2.94 |
| CC8 | -7.9 | HIS C: 280, CYS B: 240, MET B: 244 PRO B: 242, HIS B: 280 PRO C: 242, PRO B: 242 THR B: 239,HOH B: 712 |
Conventional H-Bond Carbon H-Bond Alkyl,Pi-Alkyl Unfavorable |
1.88,2.33,2.90 3.03,3.64 4.17,4.87 1.89,2.87 |
The binding interactions included various types such as hydrogen bonds, hydrophobic interactions, and π-π stacking, which are crucial for the stability of the ligand-protein complex. For instance, CC5 exhibited the highest binding affinity of-8.6 kcal/mol, indicating a stronger interaction due to multiple hydrogen bonds and significant π interactions with key residues (THR G: 37, GLY G: 38, HIS G: 43). In contrast, CC4 had a lower binding affinity (-6.8 kcal/mol) and unfavourable interactions, suggesting that its efficacy may be compromised.
Molecular dynamics
Molecular Dynamics simulations provide a comprehensive understanding of ligand-receptor interactions by accounting for receptor plasticity, which is often neglected in docking approaches. This method simulates the movement of each atom in the ligand and receptor, quantifying the forces and velocities acting on them and thus offering precise energy predictions and conformational sampling.
RMSD analysis
The RMSD analysis, depicted in fig. 4, demonstrates the stability of the protein-ligand complex throughout the 100 ns simulation period. The RMSD values for both the protein and the ligand remained consistently within the 1-3 Å range, indicating that the protein structure was largely intact, with only minor fluctuations. This stability suggests that the ligand maintains a strong and secure binding within the active site, correlating with the favourable binding affinities observed in the docking results.
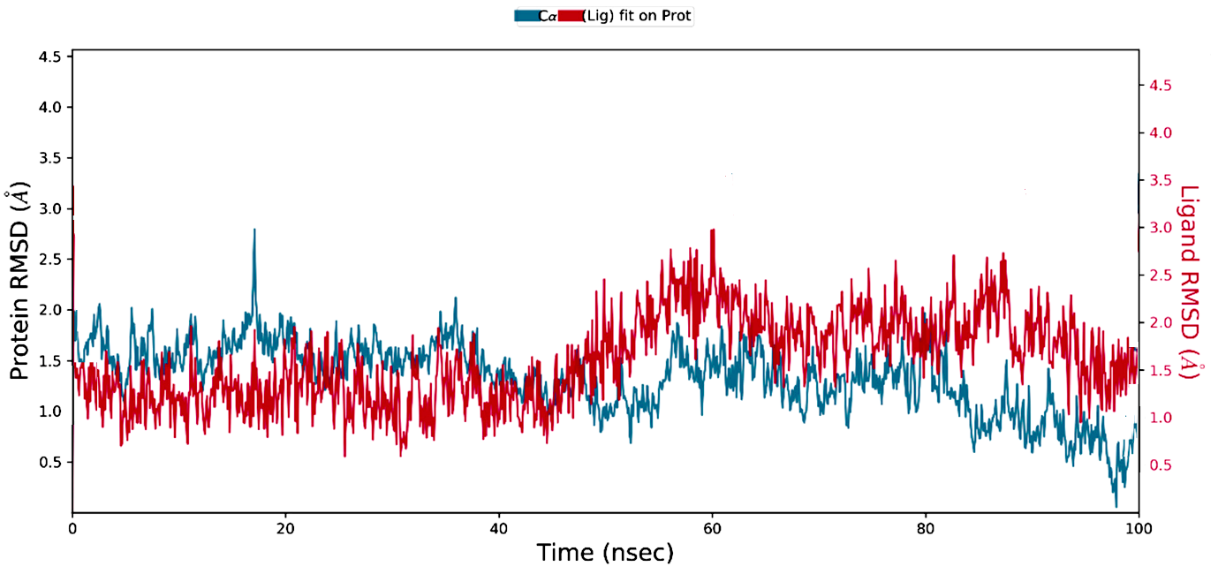
Fig. 4: Protein-ligand RMSD plot
RMSF analysis
The RMSF analysis, illustrated in fig. 5, reveals the flexibility of individual residues in the protein during the simulation. Higher RMSF values were observed for specific residues, indicating their significant conformational changes upon ligand binding. The stable interactions between the ligand and the binding site residues suggest that these fluctuations are essential for accommodating the ligand, further reinforcing the binding stability.
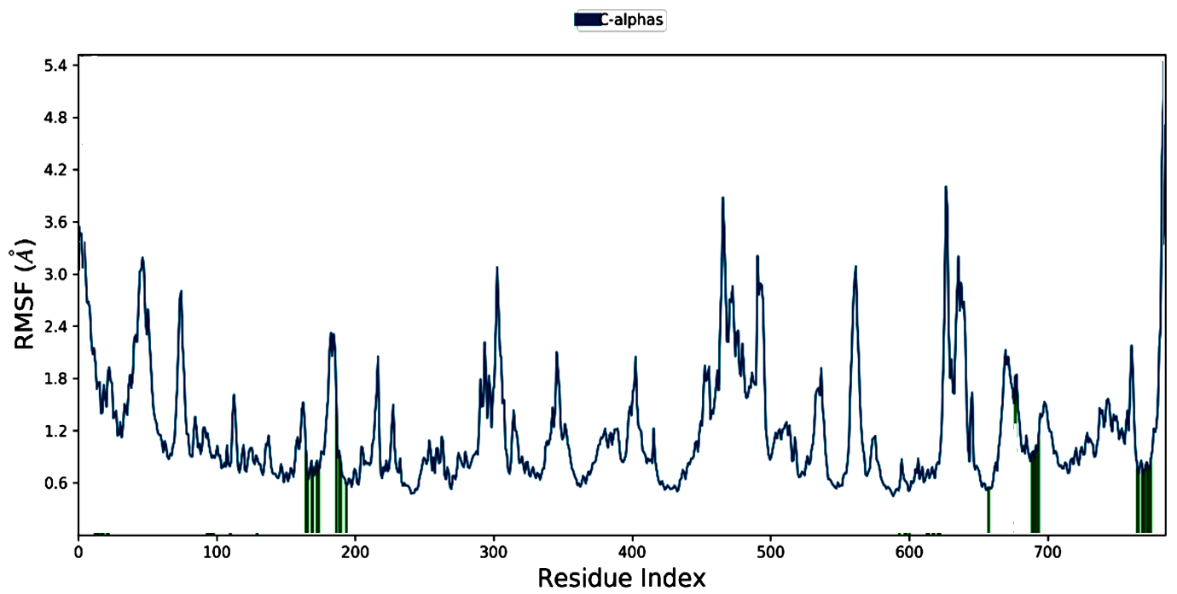
Fig. 5: RMSF plot of ligand-protein complex
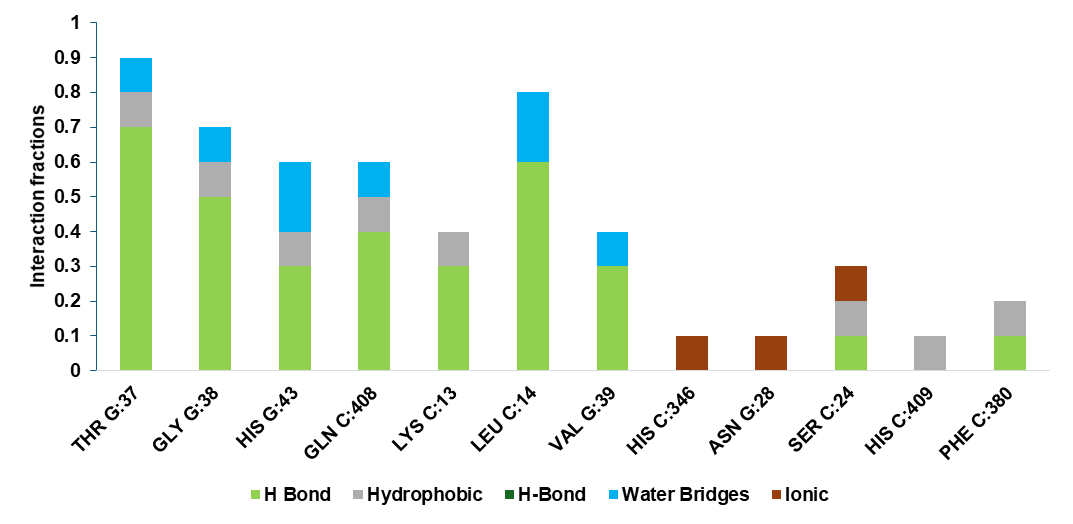
Fig. 6: Fraction of Interactions between PDB 5wb7 and compound CC5 over the 100 ns
Interaction fraction analysis
The interaction fraction analysis during the 100 ns MD simulation, shown in fig. 6, confirms the persistence of critical interactions between compound CC5 and the EGFR protein. The majority of these interactions are attributed to hydrogen bonds and water-mediated bridges, underscoring the robustness of the ligand-receptor complex. The sustained interaction fractions indicate the potential therapeutic efficacy of CC5, highlighting its suitability as an effective inhibitor in the treatment of angina pectoris.
Further analyses were conducted, including Solvent-Accessible Surface Area (SASA) plots and hydrogen bond occupancy over time. These additional metrics provided a more complete picture of ligand stability, revealing fluctuations in surface area exposure that correspond to ligand mobility and binding adaptability. The hydrogen bond occupancy analysis indicated a consistent presence of key hydrogen bonds throughout the simulation, contributing to the overall stability of the ligand-receptor complex.
SWISS ADME results
The SWISS ADME analysis offers a detailed profile of the lead compound's physicochemical properties, lipophilicity, water solubility, pharmacokinetics, and drug-likeness, essential for assessing its potential as a drug candidate. The Results of SWISS ADME were enumerated in table 5.
Physicochemical properties
The lead compound has a high number of rotatable bonds (11), indicating substantial conformational flexibility. With 8 hydrogen bond acceptors and 1 donor, it is likely to engage in strong hydrogen bonding interactions. Its molar refractivity (141.31) is typical for drug-like compounds, and the fraction of sp³-hybridized carbons (0.26) suggests a balance of rigidity and flexibility. The TPSA (123.17 Å) implies moderate polarity, affecting its solubility and absorption. The compound's moderate lipophilicity is indicated by Log Po/w values (XLOGP3: 3.84; WLOGP: 4.63), suggesting it has an appropriate balance for membrane permeability and solubility. The compound has poor water solubility with Log S values ranging from-4.83 to-6.38, which may impact its bioavailability and require formulation adjustments.
Pharmacokinetics
Pharmacokinetic predictions indicate that the compound exhibits low gastrointestinal absorption, which may restrict its oral bioavailability. Additionally, it is not anticipated to penetrate the Blood-Brain Barrier (BBB), limiting its utility for Central Nervous System (CNS) disorders but potentially advantageous for other therapeutic areas. The BOILED-Egg model, used to predict gastrointestinal absorption and brain penetration of small molecules, is illustrated in fig. 7. The red dot outside the white region of the BOILED Egg signifies the compound's low gastrointestinal absorption. The compound is not a substrate for P-glycoprotein (P-gp), suggesting that it may have improved systemic availability compared to compounds that are P-gp substrates. However, it is an inhibitor of CYP2C19, CYP2C9, and CYP2D6, which could lead to significant drug-drug interactions and impact the metabolism of co-administered drugs. The log Kp value for skin permeation is-6.58 cm/s, indicating poor absorption through the skin, which is relevant if topical formulations are considered.
Table 5: Results of SWISS ADME analysis of lead compound
| Parameter | Result |
| Num. rotatable bonds | 11 |
| Num. H-bond acceptors | 8 |
| Num. H-bond donors | 1 |
| Molar Refractivity | 141.31 |
| Fraction Csp3 | 0.26 |
| TPSA | 123.17 A° |
| Lipophilicity | |
| LogPo/w(XLOGP3) | 3.84 |
| LogPo/w(WLOGP) | 4.63 |
| Water Solubility | |
| LogS(SILICOS-IT) | -6.38 |
| LogS (ESOL) | -4.83 |
| Log S (Ali) | -6.12 |
| Pharmacokinetics | |
| GI absorption | Low |
| BBB permeant | No |
| P-gp substrate | No |
| CYP1A2inhibitor | No |
| CYP2C19inhibitor | Yes |
| CYP2C9inhibitor | Yes |
| CYP2D6 inhibitor | Yes |
| LogKp(skin permeation) | -6.58 cm/s |
| Drug likeness | |
| Lipinski | Yes; 0violation |
| Ghose | No |
| Veber | No |
| Egan | Yes |
| Muegge | Yes |
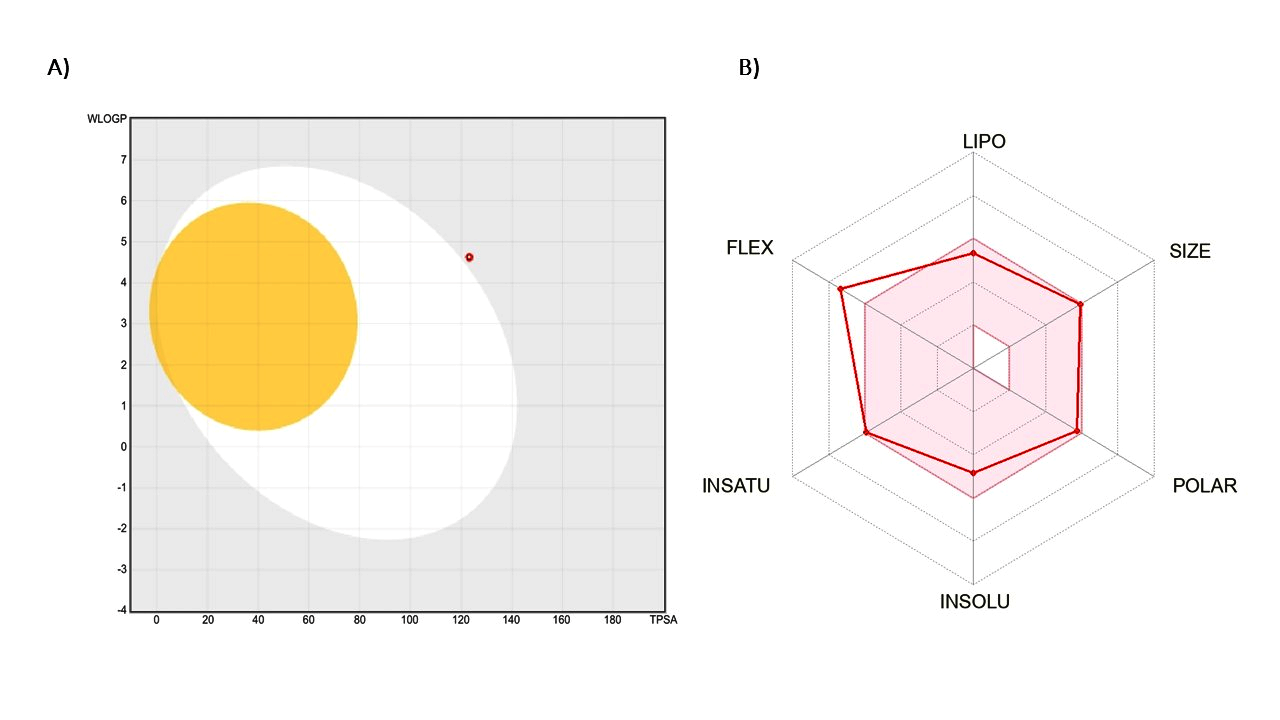
Fig. 7: Boiled egg model plot for the lead compound
Drug likeness
The compound adheres to Lipinski's Rule of Five, with zero violations, suggesting it possesses desirable drug-like properties and is likely to exhibit good oral bioavailability. However, it does not meet Ghose’s criteria, indicating potential issues with its drug-likeness profile according to this specific model. The compound also fails to meet Veber’s rules, which could suggest challenges related to oral bioavailability due to its flexibility and TPSA. On the other hand, it meets Egan’s criteria, suggesting it is likely to be orally bioavailable and have favourable pharmacokinetic properties. Additionally, the compound adheres to Muegge’s rules, supporting its potential as a drug candidate based on its physicochemical properties.
HOMO and LUMO study
The purpose of the HOMO and LUMO study is to evaluate the electronic properties of the compounds, which directly influence their chemical reactivity and stability. The HOMO energy of the parent compound was calculated to be-2.885 eV, while the LUMO energy was-1.077 eV, resulting in an energy gap of-1.808 eV. In comparison, the lead compound exhibited a HOMO value of-1.077 eV and a LUMO value of-0.507 eV, leading to a significantly smaller energy gap of-0.57 eV. The HOMO and LUMO states of the Parent compound and the lead compound were displayed in fig. 8. A smaller energy gap indicates greater stability; therefore, the lead compound, with its reduced energy gap, is more stable than the parent compound. This analysis helps in understanding the potential reactivity and stability of the compounds, guiding the selection of the lead compound for further development.
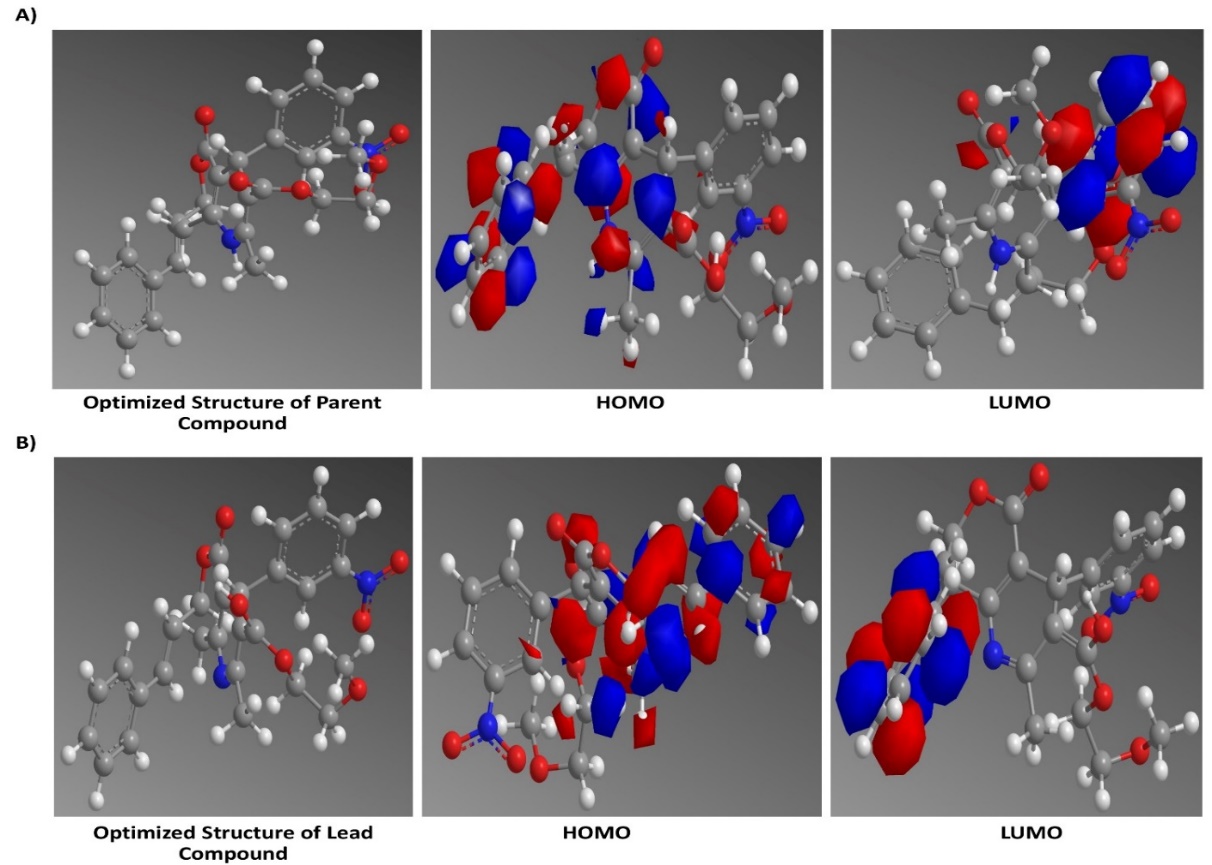
Fig. 8: A) HOMO andLUMO of the parent compound, B) HOMO and LUMO of Lead compound
Saturated solubility analysis
The solubility of the lead compound was significantly higher compared to the parent compound, as demonstrated by the saturated solubility values that are given in table 6. The parent compound exhibited a solubility of 63.33±11.53 mg/ml, while the lead compound showed a markedly increased solubility of 1198.93±11.63 mg/ml. The fold increase in solubility for the lead compound was 2.63, significantly higher than the 1.45-fold increase observed for the parent compound (P<0.001). These results indicate that the lead compound has superior solubility properties, which is a crucial factor for enhancing its bioavailability and overall therapeutic efficacy.
Table 6: Results of solubility experiments
| Compound name | Saturated solubility (mg/ml) | Folds | Significance |
| Parent Compound | 63.33±11.53 | 1.45 | P>1 |
| Lead Compound | 1198.93±11.63 | 2.63 | P<0.001 |
Data are expressed as mean±SD (n=3)
Design of experiments
The Taguchi L9 Orthogonal Array (OA) design was employed to evaluate the influence of four factors-Stabilizer Concentration, Stirring Speed, Temperature, and Solvent Ratio-on four critical responses: Particle Size, EE, Zeta Potential, and Drug Release. Each factor was tested at three levels, leading to nine experimental runs. The results were enumerated in table 7 were analysed to determine the most significant factors affecting each response, as well as the optimal conditions for nanoparticle preparation.
Particle size
The analysis of particle size using the Taguchi method revealed that stirring speed was the most significant factor, followed by temperature, stabilizer concentration, and solvent ratio. This conclusion was supported by both the response tables for means and signal-to-noise (S/N) ratios, where stirring speed had the highest Delta value (165.0) in the means table and (8.16) in the S/N ratio table. The regression analysis further confirmed the strong influence of stirring speed with a negative coefficient (-0.02063) and a significant p-value (0.034), indicating that increasing the stirring speed leads to a reduction in particle size. Temperature also showed a notable impact, with a positive coefficient (3.11), suggesting that higher temperatures increase particle size, though its p-value (0.126) was less significant. Stabilizer concentration and solvent ratio had minimal effects, as indicated by their small coefficients and high p-values. The Pareto chart (fig. 9) highlights the dominance of stirring speed and temperature in controlling particle size.
Table 7: Summary of results of taguchi model levels
| Runs | Stabilizer concentration (%) | Stirring speed (RPM) | Temperature (°C) |
Solvent: antisolvent ratio (v/v) | Particle size (nm) | EE (%) | Zeta potential (mV) | Drug release (%) |
| 1 | -1 | -1 | -1 | -1 | 258.32±1.2 | 75.21±1.4 | -28.84±0.3 | 50.35±18 |
| 2 | -1 | 0 | 0 | 0 | 250.54±1.6 | 88.64±1.3 | -36.36±0.2 | 80.23±33 |
| 3 | -1 | 1 | 1 | 1 | 180.66±5.1 | 73.97±1.5 | -35.29±0.3 | 55.61±11 |
| 4 | 0 | -1 | 0 | 1 | 315.16±1.9 | 85.07±2.1 | -53.63±0.6 | 68.94±19 |
| 5 | 0 | 0 | 1 | -1 | 208.27±1.1 | 70.34±1.3 | -51.52±0.4 | 72.57±53 |
| 6 | 0 | 1 | -1 | 0 | 50.34±3.1 | 97.09±2.1 | -48.06±0.3 | 90.46±26 |
| 7 | 1 | -1 | 1 | 0 | 330.27±2.3 | 63.63±3.4 | -58.39±0.4 | 63.27±43 |
| 8 | 1 | 0 | -1 | 1 | 130.57±1.8 | 76.34±1.5 | -45.12±0.5 | 86.39±27 |
| 9 | 1 | 1 | 0 | -1 | 178.19±1.5 | 63.61±1.3 | -36.22±0.2 | 80.27±23 |
Data are expressed as mean ±SD (n=3)
EE
For EE, stabilizer concentration emerged as the most critical factor, followed by temperature and solvent ratio, while stirring speed had the least impact [31]. The S/N ratio analysis ranked stabilizer concentration highest, with a Delta of 1.88, and the means table showed the largest difference (16.67) between levels for this factor. Regression analysis showed that stabilizer concentration had a negative effect on EE, with a coefficient of-2.83, although this was not statistically significant (P = 0.238). Temperature exhibited a negative influence on EE, as indicated by its coefficient of-0.467, though with moderate significance (p = 0.167). Solvent ratio presented varied effects depending on the levels, with the ratio 1:15 showing a reduction in EE. The Pareto chart (fig. 9) reinforces the significance of stabilizer concentration.
Zeta potential
The zeta potential analysis revealed that stabilizer concentration was the most influential factor, followed by solvent ratio, temperature, and stirring speed [21]. The response table for means showed that a lower stabilizer concentration (Level 1) yielded higher zeta potential values, whereas higher concentrations (Level 3) resulted in more negative zeta potentials, which was also supported by the S/N ratios analysis. The regression analysis showed that stabilizer concentration had a significant negative effect on zeta potential (coefficient-3.33), although the p-value (0.176) indicated that this was not highly significant [33]. The solvent ratio also impacted zeta potential, with a significant effect at 1:15. The Pareto chart (fig. 9) confirms that stabilizer concentration is the dominant factor in determining zeta potential.
Drug release
Regarding drug release, stirring speed was identified as the most influential factor, followed closely by stabilizer concentration, temperature, and solvent ratio [43]. The S/N ratio table ranked stirring speed highest (Delta 2.43), and the means table supported this with the largest difference between levels. The regression analysis, however, showed a more complex interaction, with stirring speed having a positive effect (coefficient 0.000417), though not statistically significant (P = 0.695). Stabilizer concentration also positively influenced drug release, which was supported by the higher drug release percentages at higher concentrations in the experimental runs. Temperature had a mixed impact, with the response tables indicating that moderate temperatures (Level 2) were optimal. The Pareto chart (fig. 9) emphasizes the importance of stirring speed and stabilizer concentration.
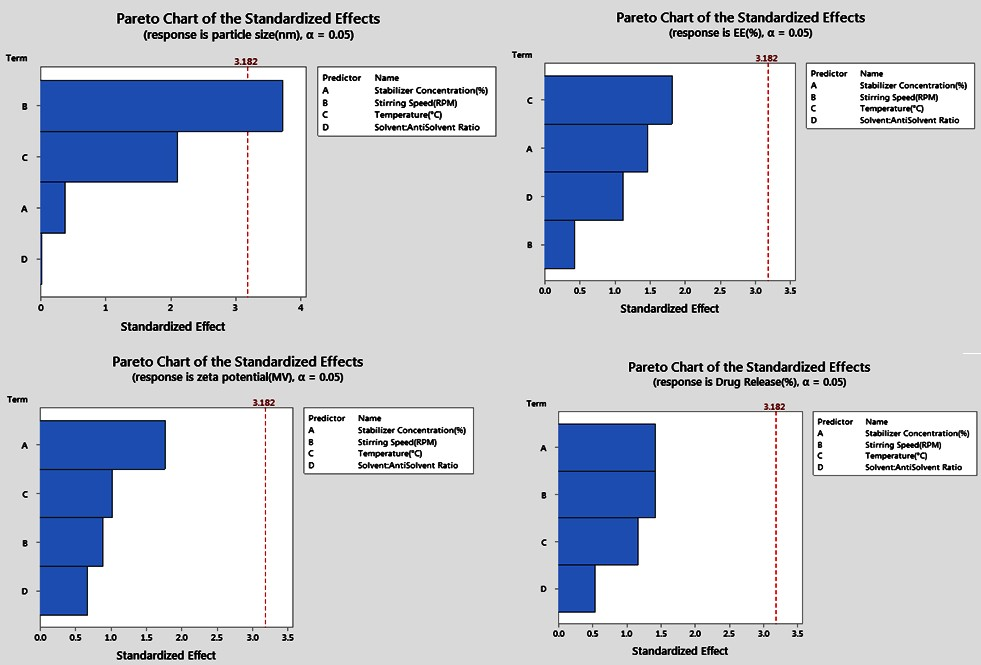
Fig. 9: Pareto chart for the response variables
Characterization of nanoparticles
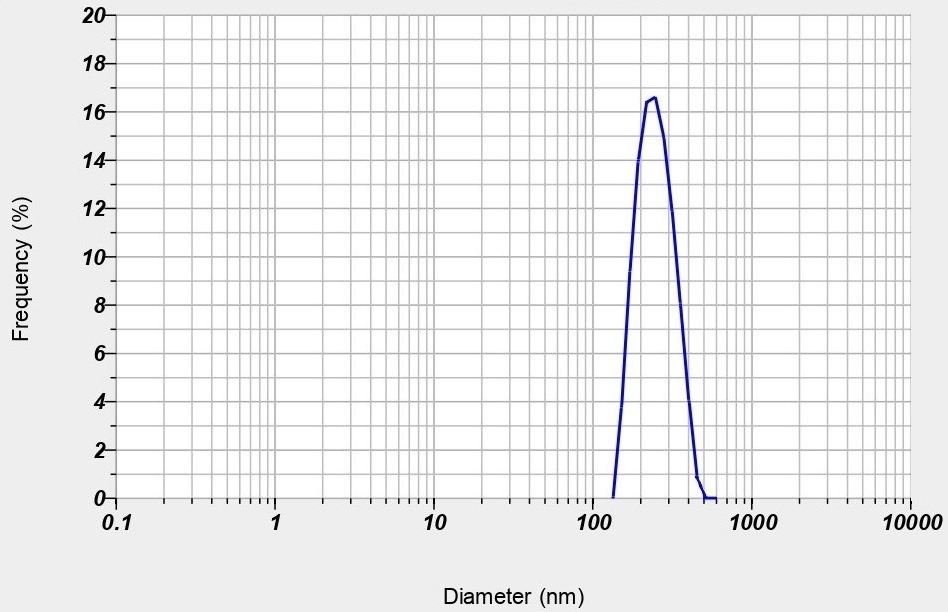
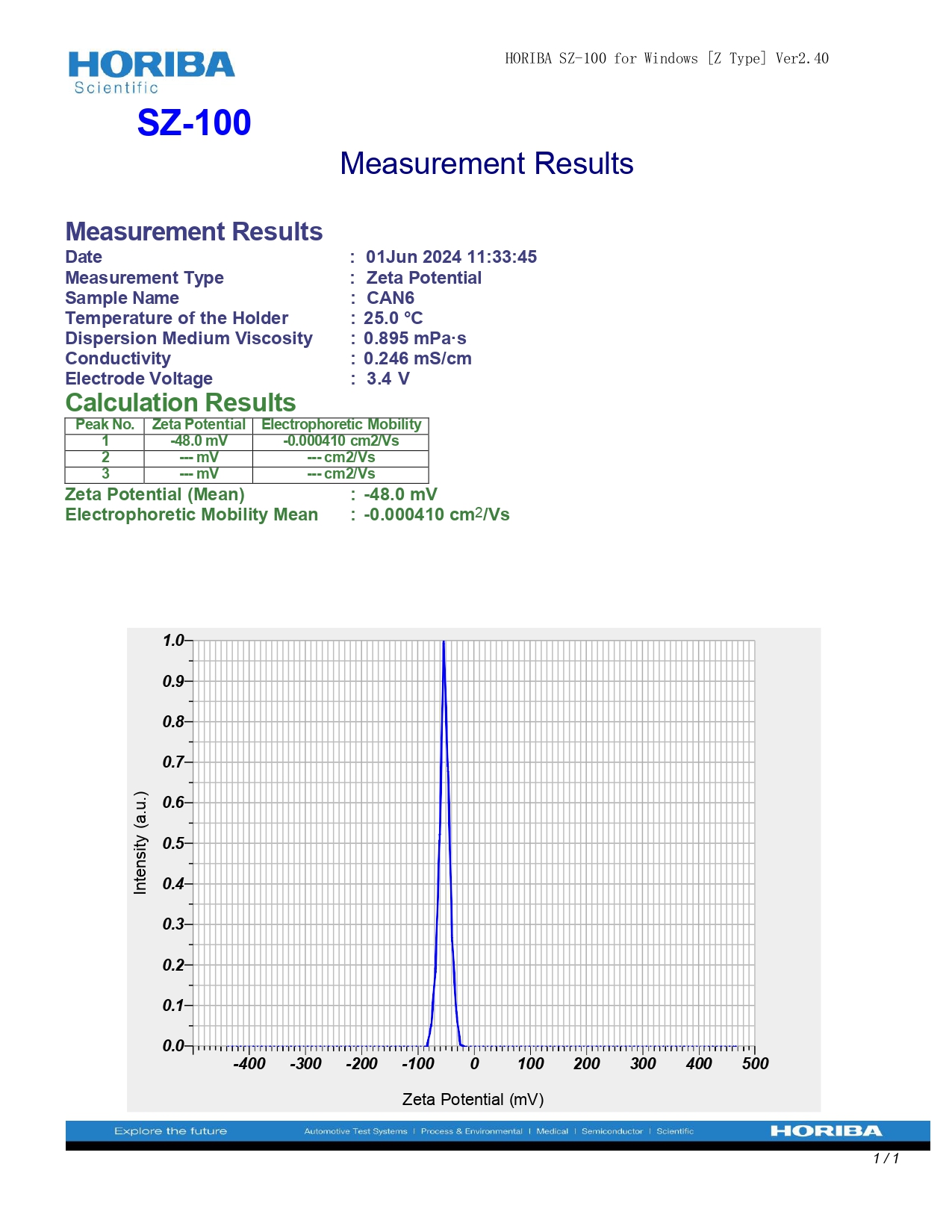
The optimized nanoparticles exhibited a particle size of 50 nm, indicating a narrow distribution favourable for enhanced cellular uptake and stability. The zeta potential measured at-48.0 mV suggests strong electrostatic repulsion between particles, which can prevent aggregation and promote colloidal stability, essential for consistent performance in biological systems. These properties, as shown in fig. 10, highlight the potential of the nanoparticles for improved bioavailability.
A)
B)
Fig. 10: A) Particle size B) Zeta potential of optimized nanoparticles
FTIR studies
In the FTIR study, no incompatibilities were observed among the different combinations tested. The FTIR spectra of Cilnidipine, the Lead Compound, Pluronic F 188, and the mixture of Lead Compound, Pluronic F 188, and Transcutol HP were analysed (fig. 11). The absence of significant shifts or new peaks in the spectra of the mixture compared to the individual components indicates that there are no chemical interactions or incompatibilities among these substances. This suggests that the components are compatible and can be used together in the formulation without any adverse effects on the stability or efficacy of the final product.
TEM study
The TEM analysis of the nanoparticles revealed detailed morphological characteristics, including their size, shape, and distribution. The nanoparticles were observed to be uniform in size with an average diameter below 265 nm can be identified from the TEM images displayed in fig. 12. They exhibited a spherical shape and were well-dispersed without significant aggregation.
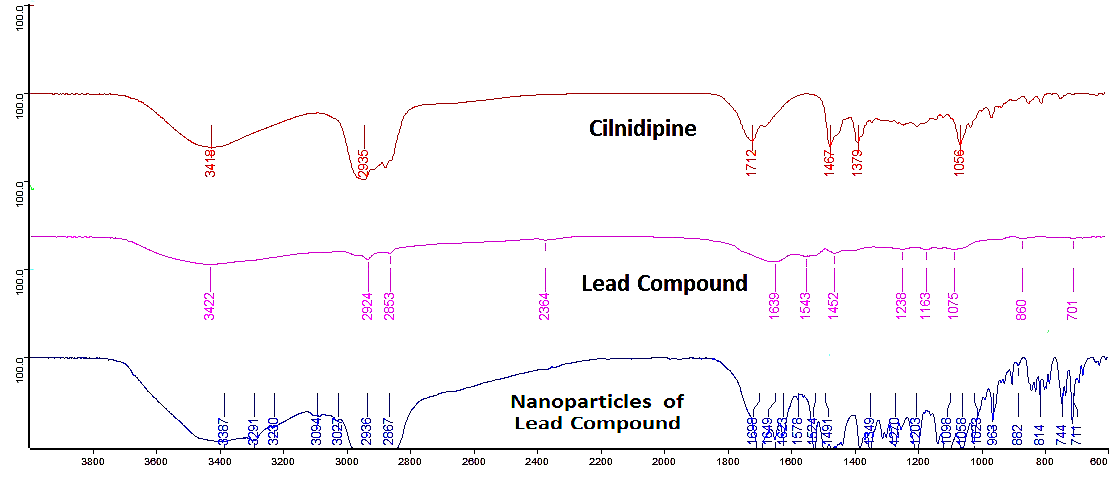
Fig. 11: FTIR of cilnidipine, lead compound, nanoparticles of lead compound

Fig. 12: TEM image of lead compound loaded nanoparticles
XRD study
The XRD studies of the Pure Drug, lead compound and its nanoparticles revealed distinct crystalline characteristics as depicted in fig. 13. The XRD pattern of the bulk Pure Drug, lead compound exhibited sharp and well-defined peaks corresponding to its crystalline structure. In contrast, the XRD pattern of the nanoparticles of Lead compound showed broader peaks with reduced intensity, indicative of a smaller crystallite size and potential partial amorphization. This shift in the diffraction pattern confirms the successful formation of nanoparticles with reduced crystallinity compared to the lead compound. The presence of characteristic peaks in both patterns confirms that the core crystalline structure of the lead compound is retained in the nanoparticulate form.
DSC
DSC studies revealed that the melting point of the lead compound was reduced to 98 °C, compared to 110 °C for pure Cilnidipine (fig. 14). In the nanoparticle formulation, the melting point of the lead compound further decreased to 90 °C (fig. 14). This reduction in thermal stability suggests that nanoparticle formation has altered the compound’s thermal behaviour, likely due to changes in crystallinity or polymorphic form. These modifications may contribute to enhanced solubility characteristics, which can improve the bioavailability of the lead compound.
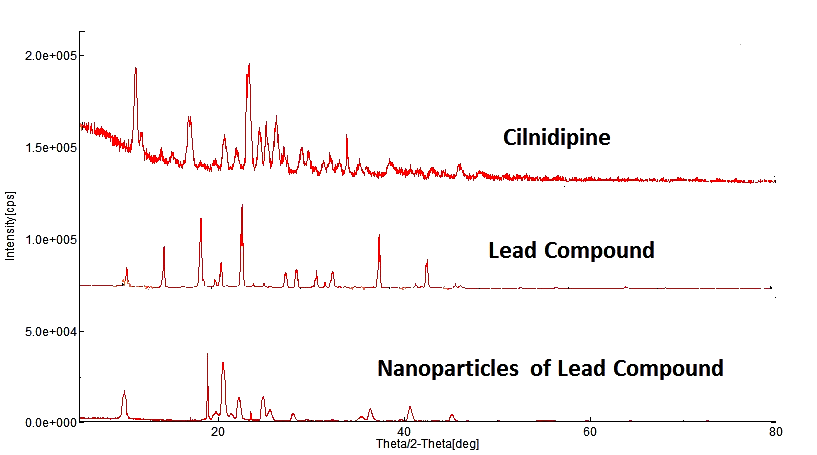
Fig. 13: XRD of cilnidipine, lead compound, nanoparticles of lead compound
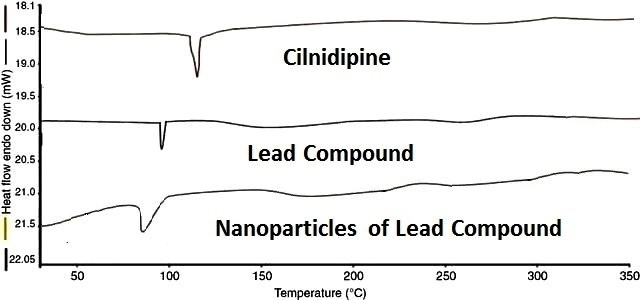
Fig. 14: DSC of cilnidipine, lead compound, nanoparticles of lead compound
Dissolution studies of cilnidipine analogue loaded nanoparticulate oral disintegrating tablets
The dissolution profiles of the Cilnidipine Analogue Loaded Nanoparticulate ODTs were assessed and compared with both controlled and marketed preparations. The results (table 8, fig. 15 and fig. 16) indicate significant differences in drug release rates among the various formulations.
Initially, at the 5-minute mark, CAF9 demonstrated the highest drug release (74.83±1.13%), which was substantially higher than the other formulations, suggesting a faster onset of action. In contrast, the marketed preparation showed no drug release at this time point, indicating a slower dissolution profile compared to all the nanoparticle formulations.
By the 10 min mark, CAF9 continued to lead with 81.67±1.53% cumulative drug release, while F7 (68.38±0.89%) also exhibited a rapid release. The controlled preparation displayed a cumulative drug release of 56.36±1.16%, which, while higher than the marketed preparation (3.35±1.05%), was still significantly lower than the nanoparticle formulations, except for CAF1.
At 20 min, CAF9 maintained its superior drug release at 87.56±1.12%, with CAF6 and CAF8 also showing high release rates of 75.37±0.13% and 74.32±1.48%, respectively. The controlled preparation lagged at 68.33±1.18%, and the marketed preparation remained far behind at 8.13±1.51%.
At the 30 min mark, CAF9 achieved almost complete drug release (98.89±1.10%), closely followed by CAF8 (91.63±1.78%), while CAF6 and CAF7 exhibited release rates around 84%. The controlled preparation reached 80.58±1.53%, whereas the marketed preparation still showed a significantly lower release of 18.85±1.01%.
The data indicate that the Cilndipine Analogue loaded nanoparticulate oral disintegrating formulations, particularly CAF9 have superior dissolution profile of 98.89% compared to both the controlled (80.58%) and marketed preparations (18.85%). This enhanced dissolution could potentially translate to improved bioavailability and therapeutic efficacy of the lead compound in clinical applications.
Table 8: % Cumulative drug release of CAF1-CAF9, controlled preparation and marketed preparation
| Time (min) | CAF1 | CAF2 | CAF3 | CAF4 | CAF5 | CAF6 | CAF7 | CAF8 | CAF9 | Controlled preparation | Marketed preparation |
| 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | 35.78±0.95 | 50.45±0.57 | 53.53±1.11 | 50.15±1.21 | 52.53±0.98 | 58.13±1.01 | 52.15±1.51 | 53.13±1.51 | 74.83±1.13 | 53.25±1.53 | 0 |
| 10 | 43.45±1.25 | 52.36±0.18 | 54.13±1.17 | 56.25±1.23 | 58.33±0.78 | 60.88±1.11 | 68.38±0.89 | 58.25±0.98 | 81.67±1.53 | 56.36±1.16 | 3.35±1.05 |
| 15 | 46.36±1.12 | 55.85±0.53 | 65.36±0.98 | 64.56±0.79 | 64.81±1.11 | 68.35±0.93 | 73.15±0.58 | 63.92±1.11 | 84.34±1.10 | 63.26±1.12 | 5.21±1.73 |
| 20 | 48.85±1.15 | 65.89±0.78 | 73.87±1.11 | 67.13±1.12 | 72.53±0.89 | 75.37±0.13 | 80.53±1.15 | 74.32±1.48 | 87.56±1.12 | 68.33±1.18 | 8.13±1.51 |
| 25 | 50.63±0.95 | 66.36±0.91 | 76.83±0.79 | 73.65±0.53 | 75.23±1.13 | 80.17±0.13 | 83.53±1.11 | 83.15±1.13 | 91.53±1.35 | 73.38±1.01 | 13.83±0.89 |
| 30 | 53.06±0.86 | 72.35±0.98 | 77.85±0.59 | 79.59±0.11 | 80.15±1.12 | 83.39±0.73 | 84.36±1.13 | 91.63±1.78 | 98.89±1.10 | 80.58±1.53 | 18.85±1.01 |
Data are expressed as mean ±SD (n=3)
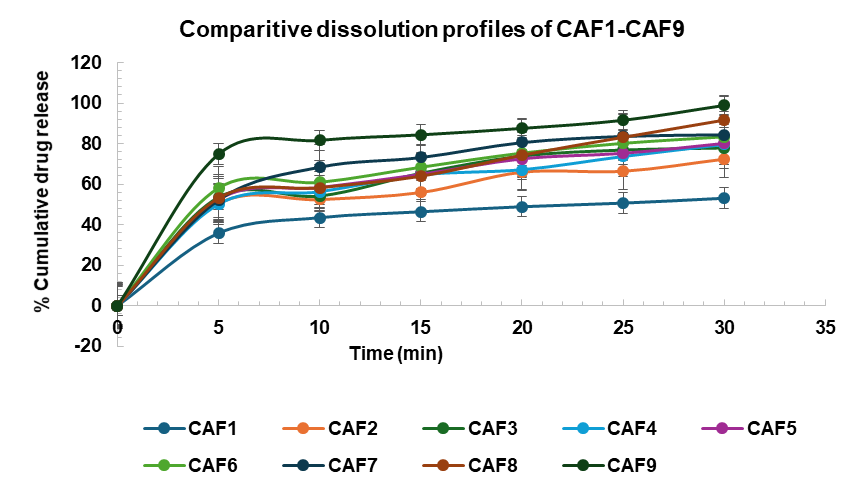
Fig. 15: Comparative dissolution profile of CAF1-CAF9, values expressed in mean±SEM (n=3)
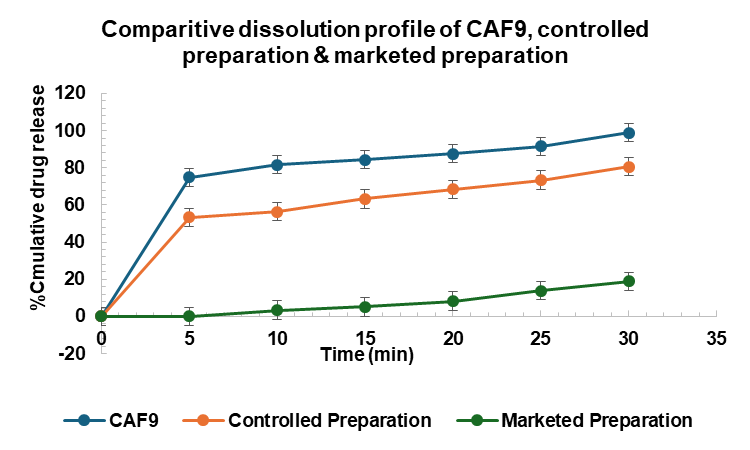
Fig. 16: Comparative dissolution profile of CAF9, controlled formulation and marketed preparation, values expressed in mean±SEM (n=3)
Stability studies of tablets
The stability studies of the optimized formulation CAF9, stored at 25 °C±2 °C/60+5%RH for 3 mo, demonstrated minimal changes in the evaluated parameters, indicating good stability under accelerated conditions. The results of stability studies were enumerated in table 9.
Regarding physical characteristics, the hardness of CAF9 remained consistent throughout the study, with values ranging from 3.55±0.24 kg/cm² after 1 mo to 3.54±0.13 kg/cm² after 3 mo, compared to the initial controlled value of 3.56±0.15 kg/cm². This consistency suggests that the formulation maintained its mechanical integrity over time.
The drug content also showed negligible variation, with an initial value of 99.08±0.07% and a slight reduction to 99.03±0.04% after 3 mo. This minimal decrease indicates that the drug remained chemically stable, with no significant degradation occurring under the storage conditions.
Table 9: Results of stability studies of optimized formulation CAF9 at 25 °C±2 °C/60+5%RH
| S. No. | Parameters | Controlled CAF9 | CAF9 | ||
| After 1 Mo | After 2 Mo | After 3 Mo | |||
| 1 | Hardness (kg/cm2) | 3.56±0.15 | 3.55±0.24 | 3.54±0.17 | 3.54±0.13 |
| 2 | Drug content (%) | 99.08±0.07 | 99.07±0.03 | 99.06±0.02 | 99.03±0.04 |
| 3 | In vitro disintegration time (sec) | 12±1.15 | 12±0.08 | 11±0.98 | 11±0.98 |
| 4 | Wetting time (sec) | 10±1.01 | 10±0.09 | 9±0.11 | 9±0.10 |
| % Cumulative drug release | |||||
| Time (min) | Controlled CAF9 | % Cumulative drug release of CAF9 | |||
| After 1 Mo | After 2 Mo | After 3 Mo | |||
| 1 | 0 | 0 | 0 | 0 | 0 |
| 2 | 5 | 74.83±1.13 | 73.54±1.10 | 73.38±1.13 | 73.28±1.11 |
| 3 | 10 | 81.67±1.53 | 81.61±1.51 | 81.59±1.31 | 81.53±1.15 |
| 4 | 15 | 84.34±1.10 | 84.23±0.58 | 90.28±1.12 | 90.13±1.13 |
| 5 | 20 | 87.56±1.12 | 87.51±0.81 | 87.45±0.19 | 87.13±0.15 |
| 6 | 25 | 91.53±1.35 | 91.50±0.89 | 91.47±0.79 | 91.11±0.56 |
| 7 | 30 | 98.89±1.10 | 98.63±1.13 | 98.48±1.15 | 98.28±1.11 |
Data are expressed as mean ±SD(n=3)
In terms of the disintegration time, the formulation exhibited a slight decrease from 12±1.15 sec at the start to 11±0.98 sec by the end of the 3 mo period. Similarly, the wetting time decreased slightly from 10±1.01 sec to 9±0.10 sec after 3 mo. These changes are minor and within acceptable limits, suggesting that the formulation's disintegration and wetting properties were largely preserved.
The in vitro dissolution profile of CAF9 over the 3 mo period also showed minimal changes. The cumulative drug release at 5 min decreased slightly from 74.83±1.13% initially to 73.28±1.11% after 3 mo. The release at 10 min remained relatively stable, with only a slight reduction from 81.67±1.53% to 81.53±1.15%. Notably, the cumulative drug release at 15 min increased slightly, reaching 90.13±1.13% after 3 mo, compared to the initial 84.34±1.10%. The release profile at 20, 25, and 30 min showed only minimal variations, with the final value at 30 min decreasing marginally from 98.89±1.10% to 98.28±1.11% after 3 mo.
The stability study results indicate that the optimized formulation CAF9 remained stable under the accelerated conditions for 3 mo, with only minor variations in hardness, drug content, disintegration time, wetting time, and dissolution profile. These findings suggest that CAF9 is likely to maintain its effectiveness and quality over an extended period, making it a promising candidate for further development and potential commercialization.
DISCUSSION
The analysis of Cilnidipine (CC1) and its derivatives, as summarized in table 3, reveals several critical pharmacokinetic and pharmacodynamic properties. All derivatives, including CC1, maintain a molecular weight of 492.58 g/mol, indicating that the structural modifications are minor and do not significantly alter the molecular framework. The log P values, ranging from 4.2758 to 5.1161, indicate varying degrees of lipophilicity. The solubility issue, with values ranging from-6.671 to-6.877, poses a challenge as poor solubility could impede drug dissolution and absorption, despite high intestinal absorption percentages (88.12% to 95.048%). Notably, the predicted solubility issues identified in silico were corroborated by in vitro dissolution studies. This discrepancy between high intestinal absorption predictions and poor in vitro dissolution underscores the limitations of computational models in fully capturing the complexities of solubility and permeability in a biological context.
The synthetic accessibility scores of 4.76 to 5.05 suggest that while the compounds are moderately challenging to synthesize, CC5and CC6, with slightly higher scores, may present additional synthesis accessibility. Toxicity predictions classify all compounds in toxicity class 5 with an LD50 of approximately 2968 mg/kg, indicating relatively low acute toxicity. Among the evaluated compounds, CC5 (ZINC101069658) and CC6 (ZINC101069663) stand out as the most promising candidates due to their balanced lipophilicity (log P of 5.11), better water solubility, and high intestinal absorption. Their manageable synthetic accessibility and favorable toxicity profile further support their potential as lead compounds for further development. CC5, in particular, is identified as the lead compound owing to its optimal balance of physicochemical properties and therapeutic potential for AP [22].
Mechanistically, CC5 exerts its effects by targeting the EGFR pathway, as confirmed by molecular docking studies. EGFR is known to play a role in cardiovascular diseases by influencing vascular smooth muscle cell proliferation and contributing to vascular remodeling, which is relevant to angina pectoris. Network pharmacology analysis further supports the involvement of EGFR and other key pathways, highlighting its potential role in modulating disease progression. The molecular docking results, showing strong binding affinity of CC5 with EGFR (PDB ID: 5wb7), were validated through MD simulations, which confirmed the stability of the protein-ligand complex. The lead compound's stable binding, as reflected by RMSD values within the 1-3 Å range, and hydrogen bond formation throughout the 100 ns simulation, suggest robust interactions that could translate into effective in vivo inhibition of EGFR-related pathways.
Comparative analysis of gene sets from OMIM, Gene Cards, Swiss Target Prediction, and STITCH databases identified a total of 47, 4592, 99, and 8 genes associated with angina pectoris, respectively. The Venn diagram highlights 60 common genes across these databases, pinpointing potential target hub genes for further investigation. A PPI network constructed from these 60 candidate genes reveals a network with 68 nodes and 186 edges, with an interaction confidence score threshold of ≥0.7. The highly interconnected nodes suggest several hub genes pivotal in the angina pectoris-related PPI network. CytoHubba plugin analysis in Cytoscape ranked the top 10 hub genes based on connectivity and centrality. SRC, though ranked first, was excluded from molecular docking due to its non-receptor tyrosine kinase nature. EGFR, the second-highest-ranking gene, was selected for docking studies due to its receptor status, making it a suitable candidate for drug-target interaction analysis [42].
Despite the promising in silico results, challenges remain in translating these findings into clinical use. Nanoparticle formulation scale-up could face difficulties related to maintaining particle uniformity and stability, particularly given the observed influence of stirring speed and stabilizer concentration on particle size and drug release. Moreover, regulatory hurdles must be addressed for novel chemical entities like CC5, especially considering the need for extensive in vivo efficacy and toxicity testing before clinical translation. The inhibition of CYP2C19, CYP2C9, and CYP2D6 by CC5 also raises concerns regarding potential drug-drug interactions, necessitating careful consideration in future clinical development.
In terms of clinical relevance, CC5’s performance must be compared to existing standards of care for angina pectoris. Currently, drugs targeting calcium channels, such as amlodipine and nifedipine, are widely used. However, CC5’s dual action of calcium channel inhibition and potential EGFR modulation offers a novel mechanism that may enhance therapeutic efficacy, particularly in patients with refractory angina. The significant reduction in the HOMO-LUMO energy gap for CC5, along with its improved solubility and promising in vitro performance, further solidifies its potential as a superior alternative to conventional therapies.
Taguchi L9 Orthogonal Array Analysis identifies stirring speed as the most significant factor affecting particle size, with temperature also having a notable impact. Increased stirring speed reduces particle size, while higher temperatures increase it. Stabilizer concentration is the most critical factor affecting EE, with a negative effect observed. Temperature also negatively influences EE, although with less significance. Stabilizer concentration significantly affects zeta potential, with higher concentrations leading to more negative values. Solvent ratio also impacts zeta potential, especially at a ratio of 1:15. Stirring speed is the primary factor influencing drug release, followed by stabilizer concentration. Higher stirring speeds enhance drug release, while stabilizer concentration also positively impacts it [44, 45].
FTIR spectra show no significant chemical interactions or incompatibilities among the components, confirming their compatibility in the formulation. TEM images reveal uniform nanoparticles with an average diameter of 50 nm, exhibiting a spherical shape and well-dispersion without aggregation. XRD patterns show distinct crystalline characteristics for the parent compound, bulk lead compound and broader peaks for nanoparticles, indicating reduced crystallinity and successful nanoparticle formation. Nanoparticle formulations, particularly CAF9, CAF8, and CAF6, demonstrate superior dissolution profiles compared to controlled and marketed preparations. CAF9, in particular, exhibits the highest drug release rates, suggesting improved bioavailability and therapeutic potential. Stability studies for formulation CAF9, stored at 25 °C±2 °C/60+5%RH for 3 mo, reveal minimal changes in hardness, drug content, disintegration time, wetting time, and dissolution profile, indicating good stability and supporting the formulation's potential for further development and commercialization.
CONCLUSION
The investigation of Cilnidipine derivatives identified CC5 (ZINC101069658) as a promising lead for AP treatment. CC5 demonstrated an optimal balance of lipophilicity, solubility, and absorption, supported by moderate log P, enhanced water solubility, and strong binding affinity with EGFR, as confirmed by molecular docking and dynamics studies. SWISS ADME and HOMO-LUMO analyses highlighted its drug-like properties and electronic stability, suggesting potential applicability in other cardiovascular conditions. Future directions include in vivo pharmacokinetic and toxicity studies to validate CC5's efficacy and safety, along with nanoparticle formulation improvements to enhance bioavailability. The Taguchi L9 design identified stirring speed and stabilizer concentration as key factors in optimizing nanoparticle stability, with formulation CAF9 showing superior drug release and stability under accelerated conditions. These findings support CC5’s advancement toward clinical application and highlight its potential in nanoparticle-based cardiovascular therapies.
ACKNOWLEDGEMENT
The authors declare their gratitude to the Department of Pharmaceutics, SRM College of Pharmacy, SRM Institute of Science and Technology, Kattankulathur, Chennai, for their encouragement and support and Santhiram College of Pharmacy, Nandyal for Providing Technical support and Suggestions from Department of Chemistry.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
Mothilal Mohan contributed to the Insilico studies, study design, while Pavankumar Krosuri collaborated with data collecting, analysis, and paper writing, assuring a collaborative and balanced effort throughout the research process. Both Authors reviewed and approved the final draft.
CONFLICT OF INTERESTS
Authors declare no conflicting interests with the present work.
REFERENCES
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001 Mar;46(1-3):3-26. doi: 10.1016/s0169-409x(00)00129-0, PMID 11259830.
Ahmmed F, Islam AU, Mukhrish YE, Bakri YE, Ahmad S, Ozeki Y. Efficient antibacterial/antifungal activities: synthesis molecular docking molecular dynamics pharmacokinetic and binding free energy of galactopyranoside derivatives. Molecules. 2022 Dec;28(1):219. doi: 10.3390/molecules28010219, PMID 36615413.
Wang M, Yang S, Shao M, Zhang Q, Wang X, LU L. Identification of potential bioactive ingredients and mechanisms of the guanxin suhe pill on angina pectoris by integrating network pharmacology and molecular docking. Evid Based Complement Alternat Med. 2021 Aug;2021:4280482. doi: 10.1155/2021/4280482, PMID 34422068.
Hosen MA, Alam A, Islam M, Fujii Y, Ozeki Y, Kawsar SM. Geometrical optimization pass prediction molecular docking and in silico admet studies of thymidine derivatives against fimh adhesion of escherichia coli. Bulg Chem Commun. 2021 Oct;53:327-42.
Kawsar SM, Hossain MA. An optimization and pharmacokinetic studies of some thymidine derivatives. Turkish Computational and Theoretical Chemistry. 2020;4(2):59-66. doi: 10.33435/tcandtc.718807.
Choudhary MI, Begum A, Abbaskhan A, Musharraf SG, Ejaz A, Atta-ur-Rahman. Two new antioxidant phenylpropanoids from Lindelofia stylosa. Chem Biodivers. 2008 Dec;5(12):2676-83. doi: 10.1002/cbdv.200890221, PMID 19089825.
Xiao ZP, Shi DH, LI HQ, Zhang LN, XU C, Zhu HL. Polyphenols based on isoflavones as inhibitors of helicobacter pylori urease. Bioorg Med Chem. 2007 Jun;15(11):3703-10. doi: 10.1016/j.bmc.2007.03.045, PMID 17400458.
LI HQ, Xiao ZP, Yin Luo T, Yan T, LV PC, Zhu HL. Amines and oximes derived from deoxybenzoins as helicobacter pylori urease inhibitors. Eur J Med Chem. 2009 May;44(5):2246-51. doi: 10.1016/j.ejmech.2008.06.001, PMID 18625539.
Xiao ZP, Wang XD, Peng ZY, Huang S, Yang P, LI QS. Molecular docking kinetics study and structure activity relationship analysis of quercetin and its analogous as helicobacter pylori urease inhibitors. J Agric Food Chem. 2012 Oct;60(42):10572-7. doi: 10.1021/jf303393n, PMID 23067328.
Xiao ZP, Peng ZY, Dong JJ, HE J, Ouyang H, Feng YT. Synthesis structure activity relationship analysis and kinetics study of reductive derivatives of flavonoids as helicobacter pylori urease inhibitors. Eur J Med Chem. 2013 May;63:685-95. doi: 10.1016/j.ejmech.2013.03.016, PMID 23567958.
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT. Glide: a new approach for rapid accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004 Mar;47(7):1739-49. doi: 10.1021/jm0306430, PMID 15027865.
Sareen S, Mathew G, Joseph L. Improvement in solubility of poor water soluble drugs by solid dispersion. Int J Pharm Investig. 2012 Jan;2(1):12-7. doi: 10.4103/2230-973X.96921, PMID 23071955.
Bhalani DV, Nutan B, Kumar A, Singh Chandel AK. Bioavailability enhancement techniques for poorly aqueous soluble drugs and therapeutics. Biomedicines. 2022 Aug;10(9):2055. doi: 10.3390/biomedicines10092055, PMID 36140156.
Kumari L, Choudhari Y, Patel P, Gupta GD, Singh D, Rosenholm JM. Advancement in solubilization approaches: a step towards bioavailability enhancement of poorly soluble drugs. Life (Basel). 2023 Apr;13(5):1099. doi: 10.3390/life13051099, PMID 37240744.
Khan KU, Minhas MU, Badshah SF, Suhail M, Ahmad A, Ijaz S. Overview of nanoparticulate strategies for solubility enhancement of poorly soluble drugs. Life Sci. 2022 Feb;291:120301. doi: 10.1016/j.lfs.2022.120301, PMID 34999114.
Abuzar SM, Hyun SM, Kim JH, Park HJ, Kim MS, Park JS. Enhancing the solubility and bioavailability of poorly water soluble drugs using supercritical antisolvent (SAS) process. Int J Pharm. 2018 Mar;538(1-2):1-13. doi: 10.1016/j.ijpharm.2017.12.041, PMID 29278733.
Chakraborty RN, Langade D, More S, Revandkar V, Birla A. Efficacy of cilnidipine (L/N-type calcium channel blocker) in treatment of hypertension: a meta-analysis of randomized and non randomized controlled trials. Cureus. 2021 Nov;13(11):e19822. doi: 10.7759/cureus.19822, PMID 34963839.
Wang AL, Iadecola C, Wang G. New generations of dihydropyridines for treatment of hypertension. J Geriatr Cardiol. 2017 Jan;14(1):67-72. doi: 10.11909/j.issn.1671-5411.2017.01.006, PMID 28270844.
Alviar CL, Devarapally S, Nadkarni GN, Romero J, Benjo AM, Javed F. Efficacy and safety of dual calcium channel blockade for the treatment of hypertension: a meta-analysis. Am J Hypertens. 2013 Feb;26(2):287-97. doi: 10.1093/ajh/hps009, PMID 23382415.
Tingle BI, Tang KG, Castanon M, Gutierrez JJ, Khurelbaatar M, Dandarchuluun C. Zinc-22-A free multi-billion scale database of tangible compounds for ligand discovery. J Chem Inf Model. 2023 Apr;63(4):1166-76. doi: 10.1021/acs.jcim.2c01253, PMID 36790087.
LE TT, DO PC. Molecular docking study of various enterovirus-A71 3C protease proteins and their potential inhibitors. Front Microbiol. 2022 Sep;13:987801. doi: 10.3389/fmicb.2022.987801, PMID 36246267.
Khalid Z, Shafqat SS, Ahmad HA, Munawar MA, Mutahir S, Elkholi SM. A combined experimental and computational study of novel benzotriazinone carboxamides as alpha glucosidase inhibitors. Molecules. 2023 Sep;28(18):6623. doi: 10.3390/molecules28186623, PMID 37764399.
Duran Iturbide NA, Diaz Eufracio BI, Medina Franco JL. In silico ADME/Tox profiling of natural products: a focus on biofacquim. ACS Omega. 2020 Jun;5(26):16076-84. doi: 10.1021/acsomega.0c01581, PMID 32656429.
Pires DE, Blundell TL, Ascher DB. Pkcsm: predicting small molecule pharmacokinetic and toxicity properties using graph based signatures. J Med Chem. 2015 May;58(9):4066-72. doi: 10.1021/acs.jmedchem.5b00104, PMID 25860834.
Amberger JS, Hamosh A. Searching online mendelian inheritance in man (OMIM): a knowledgebase of human genes and genetic phenotypes. Curr Protoc Bioinformatics. 2017 Jun;58:1.2.1-1.2.12. doi: 10.1002/cpbi.27, PMID 28654725.
Tanoli Z, Seemab U, Scherer A, Wennerberg K, Tang J, Vaha Koskela M. Exploration of databases and methods supporting drug repurposing: a comprehensive survey. Brief Bioinform. 2021 Feb;22(2):1656-78. doi: 10.1093/bib/bbaa003, PMID 32055842.
Trott O, Olson AJ. Auto dock vina: improving the speed and accuracy of docking with a new scoring function efficient optimization and multithreading. J Comput Chem. 2010 Feb;31(2):455-61. doi: 10.1002/jcc.21334, PMID 19499576.
Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure based drug discovery. Curr Comput Aided Drug Des. 2011 Jun;7(2):146-57. doi: 10.2174/157340911795677602, PMID 21534921. PMCID PMC3151162.
Daina A, Michielin O, Zoete V. Swiss ADME: a free web tool to evaluate pharmacokinetics drug likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017 Mar;7:42717. doi: 10.1038/srep42717, PMID 28256516.
Mumit MA, Pal TK, Alam MA, Islam MA, Paul S, Sheikh MC. DFT studies on vibrational and electronic spectra HOMO-LUMO MEP HOMA NBO and molecular docking analysis of benzyl-3-N-(2,4,5-trimethoxyphenylmethylene) hydrazinecarbodithioate. J Mol Struct. 2020 Nov;1220:128715. doi: 10.1016/j.molstruc.2020.128715, PMID 32834109.
Budiman A, Aulifa DL. A comparative study of the pharmaceutical properties between amorphous drugs loaded mesoporous silica and pure amorphous drugs prepared by solvent evaporation. Pharmaceuticals (Basel). 2022 Jun 9;15(6):730. doi: 10.3390/ph15060730, PMID 35745649, PMCID PMC9228546.
El-Assal MI, Samuel D. Optimization of rivastigmine chitosan nanoparticles for neurodegenerative alzheimer; in vitro and ex vivo characterizations. Int J Pharm Pharm Sci. 2022 Jan;14(1):17-27. doi: 10.22159/ijpps.2022v14i1.43145.
Madan JR, Khude PA, Dua K. Development and evaluation of solid lipid nanoparticles of mometasone furoate for topical delivery. Int J Pharm Investig. 2014 Feb;4(2):60-4. doi: 10.4103/2230-973X.133047, PMID 25006550.
Kamaraj N, Rajaguru PY, Issac PK, Sundaresan S. Fabrication characterization in vitro drug release and glucose uptake activity of 14-deoxy 11, 12-didehydroandrographolide loaded polycaprolactone nanoparticles. Asian J Pharm Sci. 2017 Jul;12(4):353-62. doi: 10.1016/j.ajps.2017.02.003, PMID 32104346.
Mhetre RL, Hol VB, Chanshetti RR, Dhole SN. Optimisation of cilnidipine nanoparticles using box-behnken design: in vitro toxicity and bioavailability assessment. Materials Technology. 2022;37(11):1796-807. doi: 10.1080/10667857.2021.1988038.
Kumar JA, Bhikshapathi DV. Development of nilotinib loaded solid lipid nanoparticles and optimization by central composite design approach. Int J App Pharm. 2022 Mar;14(2):172-80. doi: 10.22159/ijap.2022v14i2.43943.
Mourdikoudis S, Pallares RM, Thanh NT. Characterization techniques for nanoparticles: comparison and complementarity upon studying nanoparticle properties. Nanoscale. 2018 Jan;10(27):12871-934. doi: 10.1039/c8nr02278j, PMID 29926865.
Nagai N, Ogata F, Kadowaki R, Deguchi S, Otake H, Nakazawa Y. Orally disintegrating tablets containing famotidine nanoparticles provide high intestinal absorbability via the energy dependent endocytosis pathway. Front Bioeng Biotechnol. 2023 Mar;11:1167291. doi: 10.3389/fbioe.2023.1167291, PMID 36970629.
Viswaja M, Bhikshapathi DV, Palanati M, Babu AK, Goje A. Formulation and evaluation of ibrutinib nanosponges incorporated tablet. Int J Appl Pharm. 2023 Mar;15(2):92-7.
Alhalaweh A, Alzghoul A, Mahlin D, Bergstrom CA. Physical stability of drugs after storage above and below the glass transition temperature: relationship to glass forming ability. Int J Pharm. 2015 Jan;495(1):312-7. doi: 10.1016/j.ijpharm.2015.08.101, PMID 26341321.
Kurian T. Molecular docking study of epigallocatechin gallate on FLT3 in complex with gilteritinib for anticancer activity. Asian J Pharm Clin Res. 2024 Jan;17(1):5-7. doi: 10.22159/ajpcr.2024.v17i1.48733.
Chakrabarti M. Revolutionizing antimicrobial drug discovery: computational design and ADMET studies of emerging potent anti-microbial agents. Int J Pharm Pharm Sci. 2023 Aug;15(8):28-35. doi: 10.22159/ijpps.2023v15i8.48526.
Aiswariya, BVS, Satya MS. Molecular docking and admet studies of benzotriazole derivatives tethered with isoniazid for antifungal activity. Int J Curr Pharm Sci. 2022;14(4):78-80. doi: 10.22159/ijcpr.2022v14i4.2004.
Pant A, Sharma G, Saini S, Kaur G, Jain A, Thakur A. QBD driven development of phospholipid embedded lipidic nanocarriers of raloxifene: extensive in vitro and in vivo evaluation studies. Drug Deliv Transl Res. 2024 Mar;14(3):730-56. doi: 10.1007/s13346-023-01427-3, PMID 37768530.
Birla D, Khandale N, Bashir B, Shahbaz Alam M, Vishwas S, Gupta G. Application of quality by design in optimization of nanoformulations: principle perspectives and practices. Drug Deliv Transl Res. 2024 Aug 10. doi: 10.1007/s13346-024-01681-z, PMID 39126576.