
Int J App Pharm, Vol 17, Issue 2, 2025, 393-401Original Article
IN SILICO INVESTIGATION OF SMALLANTHUS SONCHIFOLIUS COMPOUNDS AS DPP-4 INHIBITORS FOR ANTIDIABETIC MECHANISMS
NOVI YANTIH, ZUHELMI AZIZ, ESTI MUMPUNI, NURUL WIDAYANTI, ANDRI PRASETIYO*
Department of Pharmaceutical Science, Faculty of Pharmacy, Pancasila University, South Jakarta, Jakarta, Indonesia
*Corresponding author: Andri Prasetiyo; *Email: andriprasetiyo@univpancasila.ac.id
Received: 22 Sep 2024, Revised and Accepted: 21 Dec 2024
ABSTRACT
Objective: Smallanthus sonchifolius has been scientifically demonstrated to possess antidiabetic activity through the inhibition of DPP-4 in in vitro studies. However, there is still a lack of comprehensive research identifying the specific bioactive compounds responsible for this effect. This research specifically aims to explore the potential of bioactive compounds from Smallanthussonchifoliusas DPP-4 inhibitors, a known target for antidiabetic treatment, using in silico techniques.
Methods: The methodologies employed in this study includedmolecular docking, ADMET prediction, and molecular dynamics simulation. The docking process was conducted on 20 test compounds against native ligands within the receptor designated by code 3G0B, as well as against comparative compounds.
Results: Molecular docking analysis revealed four compounds—3,4-dicaffeoylquinic acid, Nystose, 1,3-O-dicaffeoylquinic acid, and 3,5-caffeoylquinic acid-that exhibited lower rerank scores than the positive control, alogliptin. Further investigation through molecular dynamics simulations demonstrated that the Nystose-ligand complex displayed stable binding dynamics similar to alogliptin, maintaining consistent interactions throughout the simulation. Key amino acid residues, including Glu205, Glu206, Ser209, Tyr547, Tyr662, and Ser630, were involved in critical hydrogen bonding, contributing to the stability of the Nystose complex. However, despite its promising binding profile, Nystose is predicted to have limited intestinal absorption due to the high number of polar substituents, which may impact its bioavailability.
Conclusion: Nystose is predicted to act as a DPP-4 inhibitor for diabetes treatment based on findings from molecular docking and molecular dynamics simulations.
Keywords: Smallanthus sonchifolius, In silico, DPP-4 inhibitor
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i2.52741 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Diabetes mellitus is defined as a state of persistent hyperglycemia (elevated blood glucose) and existing metabolic disturbances affecting carbohydrates, proteins, and lipids, which are caused by deficiency in insulin secretion or action or both [1]. Diabetes today represents a major worldwide challenge to health by extremely high and lifting prevalence, difficulties in successful treatment of diabetes accompanied with huge associated morbidity. Of antidiabetic agents, DPP-4 inhibitors deserve to be considered as having higher anti-diabetes performance/safety ratio over other antihyperglycemics mainly due its lower hypoglycaemia risk [2, 3]. In addition, DPP-4 inhibitors have been particularly proven to be beneficial in patients with chronic kidney disease [4]. However, their relatively high cost compared to other antidiabetic medications makes them very expensive and necessitates more affordable plant-based alternatives. To date, DPP-4 inhibitors sourced from natural products have not been fully explored or identified.
Yacon (Smallanthus sonchifolius) has traditionally been used in ethnomedicine due to its putative antidiabetic properties, showing good promise as a natural therapeutic agent against this metabolic disease [5]. Previously, we reported that the 95% ethanol extract of yacon leaves at a concentration of 2.5 ppm exhibited strong potential for antidiabetic activity in an in vitro investigation; DPP-4 inhibition approximated 52.84% [6]. In vivo studies using ethanol extract of leaves from yacon showed that gavaged for 14 d at a dose of 300 mg/Kg body weight, it significantly decreased by 29 % the blood glucose levels in diabetic rats. Similarly, whereas a higher dose at 400 mg/Kg body weight resulted in the most significant reduction of glucose level (59%), this underscores that yacon leaf extract might be administrated as an alternative prophylaxis for controlling glycemic condition [7, 8]. The in vivo studies cannot explain the mechanism involved with glucose-lowing effects. According to the results, there is a need for further research to discover unique active compounds that have DPP-4 inhibitory activity as shown using in silico studies of yacon.
In silico techniques have found an application in the drug discovery and development paradigm, superseding traditional pharmaceutical research to a much-exalted level [9]. By utilizing algorithms such as molecular docking and molecular dynamics, researchers can accurately forecast the binding poses of prospective drug molecules with biological targets [10]. Such methods help to quickly discover lead compounds and hence shorten the pathway from identification of drug candidates to eventual product delivery while reducing costs and time. While in silico methods provide several important benefits for drug discovery and development, there are limitations of these approaches. However, in silico models fail at capturing the real complexity of biological systems and environments, which limit their accuracies to correctly predict what is going on from a computational point of view. Therefore, in silico study results must be firmly validated using the classical laboratory-based methods to secure their reliability and translational potential [11].
Molecular docking was performed in order to predict the binding of potential drug molecules with specified target against using Molegro Virtual Docker [12]. This software used to do rigorous binding affinites and, when bound how ligand can be oriented into the target protein site, which directly help us understand medicinal chemistry of drugs. Additionally, all the protein-ligand complexes were subjected to molecular dynamics simulations using YASARA Dynamics for analyzing the stability and conformation of binding poses over time. Given these computational techniques together with molecular simulations, the modeling of macromolecular complexes and cellular pathways will confer a detailed insight into dynamic interactions in biological context to drive more targeted design for the next generation therapeutics.
The aim of this work is to investigate the bioactivity of compounds extracted from yacon leaves through in silico methods. In silico techniques, molecular docking and molecular dynamic simulation will be conducted thorough this study to recognize the probable uses of these compounds as DPP-4 inhibitors. This study seeks to aid in understanding the mechanism of action and therapeutic potential by enlightening binding affinities interactions with compounds within active site DPP-4 using these computational techniques.
MATERIALS AND METHODS
The 20 test ligands used were compounds in the plants Smallanthus sonchifolius. The comparator ligands were sitagliptin and alogliptin, receptors of the DPP-4 enzyme (PDB codes: 3G0B and 5Y7K). The selection of the target proteins 3G0B and 5Y7K was meticulously aligned with stringent criteria to ensure the reliability and relevance of the molecular docking and dynamic simulations [13]. First, both proteins exhibit a resolution below 2.5 Å, a benchmark indicating a high level of structural detail essential for precise modeling of protein-ligand interactions. The structural resolution below this threshold allows for the accurate identification of binding sites and the evaluation of ligand fit within the active site. Moreover, both target proteins are derived from Homo sapiens, ensuring that the findings are directly applicable to human biology, which is critical for drug discovery and development. The inclusion of native ligands, which are existing marketed drugs, further strengthens the biological and pharmaceutical relevance of the study, as these ligands provide a validated comparison for assessing the binding efficiency and potential therapeutic viability of the test compounds. Importantly, both proteins are wild-type forms with no reported mutations, maintaining their native conformation and function. This is crucial for ensuring the integrity of the molecular simulations, as mutations can alter the protein’s binding site and overall dynamics, potentially leading to inaccurate predictions. By selecting proteins that meet these criteria, this study aims to produce robust and biologically meaningful insights into the inhibitory potential of the test compounds. Laptop, Lenovo Ideapad Slim 7 Windows 11 Home 64-bit. Intel I5 10300H with GPU NVIDIA GTX 1650 RAM 16 GB Memory DDR4 16GB, Microsoft Windows 11 Pro 64-bit, Molegro Virtual Docker (MVD) 5.0, ChemBioDraw Professional 16.0, ChemBio3D Professional (PerkinElmer Inc. Cambridge, MA, USA), Protein Data Bank (https://www.rcsb.org/), PubChem (https://pubchem.ncbi.nlm.nih.gov/), BIOVIA DiscoveryStudio v2024, pkCSM (https://biosig.lab.uq.edu.au/pkcsm/), YASARA Dynamics, Graphpad Prism 10, In the molecular dynamics simulations conducted for this study, specific parameters were systematically adjusted to optimize the simulation conditions for better biological relevance. The simulation temperature was increased from the default value of 298 K to 310 K to replicate human physiological conditions more accurately. Additionally, the simulation duration was capped at 50,000 picoseconds, allowing for a sufficiently detailed observation of molecular interactions within a finite timespan. The snapshot save interval was also refined from 100,000 femtoseconds to 10,000 femtoseconds, enhancing the frequency of data capture to ensure a more granular analysis of molecular behavior throughout the simulation. These parameter modifications were critical in ensuring that the simulated molecular dynamics more closely mirrored real-world biological environments, providing a more robust and precise assessment of the stability and functional behavior of the molecular complexes under study.
Internal validation
Molegro Virtual Docker (MVD) was utilized to perform the redocking of native ligands in the crystal structures of DPP-4 enzyme (PDB codes 3G0B and 5Y7K). The MVD software was configured automatically, ensuring proper hydrogen atoms were added, missing bonds were corrected, partial charges were assigned where needed, and flexible torsions were applied to the detected ligands. Five potential binding cavities were identified within the enzyme structures; however, only one cavity was chosen for in-depth docking analysis. A total of twelve combinations comprising three docking algorithms and four scoring functions were tested. The docking protocol was selected based on the protein model yielding the lowest RMSD value, ideally nearing zero. A protocol is deemed valid when the RMSD value is below 2 Å [14], as this indicates a close correspondence between the predicted docking pose and the experimentally derived native ligand conformation. Such accuracy implies that the predicted binding mode and ligand orientation within the active site are highly reliable. Achieving an RMSD value below 2 Å is crucial for validating docking accuracy, as it suggests that the computational predictions are reflective of actual biological interactions. In contrast, higher RMSD values signal substantial deviation from the native ligand structure, potentially leading to erroneous interpretations regarding ligand binding affinity, efficacy, and drug design prospects. Therefore, an RMSD threshold of less than 2 Å is widely accepted as a benchmark for validating docking results in structure-based drug discovery.
Preparation of test/comparator compounds in 3D structure
The 3D structure preparation for the 20 test compounds, previously identified from Smallanthus sonchifolius also by other researchers, as well as the comparator compounds sitagliptin and alogliptin, was performed using standard molecular modeling techniques. All compound structures were downloaded from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/) in the form of 2D chemical structures. These structures were then imported into the Chem3D 16.0 software for further processing. Since the initial structures obtained from the database are often not in their most energetically favorable conformations, energy minimization was carried out using the MMFF94 (Merck Molecular Force Field 94) algorithm. This step ensures that the molecules adopt their most stable 3D geometry, which is essential for accurate molecular docking and dynamic simulations. Once the structures were optimized, they were saved in the MDL Molfile format (*.mol) for subsequent analysis.
Docking molecular
A total of 20 test compounds and comparator compounds were simultaneously docked in the crystal structure of the selected DPP-IV enzyme (PDB code 3G0B). Site region binding sites of bound crystallographic ligands with sizes of X-axis: 42.21 Å, Y: 34.47 Å, and Z: 14,97 Å. Molecular docking uses the default settings of MVD, which is the PLANTS (Grid) Score algorithm and scoring function Moldock SE. Docking was performed in four replicates and run with the best combination of PLANTS (Grid) Score algorithm and scoring function MolDock SE.
Visualization of docking results
The BIOVIA Discovery studio program was used to visualize the docking results. Ligands in test compounds that interact with amino acid residues and form bonds similar to ligands in comparator compounds indicate similar activity because they involve the same type of interaction.
ADMET prediction analysis of test ligand
ADMET predictions were performed on test ligands from the best re-rank score results downloaded on the page https://pubchem.ncbi.nlm.nih.gov/in SMILES format. The prediction was made by uploading the selected ligand on the https://biosig.lab.uq.edu.au/pkcsm/ website.
Molecular dynamics simulation
Combined preparation of the 3G0B receptor and the best test ligand, which is 3,4-Dicaffeoylquinic acid, Nystose, and 1,3-O-Dicaffeoylquinic acid is done with PyMol first and then analyzed with YASARA-Structure. Simulation using the macro “md_run. mcr” with a selection of time of 50 ns, the addition of "AddSpring", and every 10 ps, the simulation trajectory is saved. The cell physics was set at 310 K, pH 7.4, and NaCl 0,9%. Analysis of simulation results using “md_analyze. mcr” in YASARA-Structure. The selected analysis is the calculation of Root mean Square Deviation (RMSD), Root mean Square Fluctuation (RMSF), and binding energy. The analysis results were saved in the form of a notepad file and processed using Microsoft Excel, and the graph was formed using the GraphPad Prism 10 program.
RESULTS
Internal validation (Redocking) of proteins
The redocking procedure aims to validate the accuracy of the docking protocol by ensuring its capability to reproduce the precise positioning of the native ligand within the binding site. This is achieved by comparing the docked pose of the native ligand with its original position in the co-crystal structure (before docking). The internal validation (redocking) conducted on two proteins (PDB codes 3G0B and 5Y7K), as presented in tables 1 and 2, using Molegro Virtual Docker (MVD), yielded significant results. The lowest RMSD value of 0.383922 Å was achieved with the 3G0B protein, utilizing the Moldock SE search algorithm combined with the plant score grid scoring function. The binding site coordinates for this protein were determined as X = 42.21 Å, Y = 34.47 Å, and Z = 14.97 Å. This observation aligns with prior research demonstrating that MolDock successfully identifies the correct binding modes in 87% of protein-ligand complexes, showcasing its robust predictive capabilities in molecular docking applications. As illustrated in fig. 1a, this RMSD value, which is below 2 Å, indicates that the docking protocol accurately positioned the native ligand in nearly the same pose as before docking, signifying that the protocol effectively replicates true biological interactions. In contrast, fig. 2b demonstrates an RMSD value greater than 2 Å, suggesting that the docking protocol failed to reproduce the native ligand's original binding position. This discrepancy could lead to misinterpretation regarding the ligand's binding affinity and biological relevance, highlighting the importance of accurate docking validation to avoid errors in structure-based drug discovery. Based on the findings, protein 3G0B was selected for further investigation, employing the Moldock SE docking algorithm in conjunction with the Plant Score (grid) scoring function. The validation results indicate that the docking protocol is reliable and suitable for use in future predictive analyses. This methodology aims to enhance the reliability of predictions regarding ligand-receptor interactions, thereby contributing to the identification of potential therapeutic agents. Such an approach aligns with established practices in structure-based drug design, where accurate docking simulations are crucial for understanding binding affinities and optimizing lead compounds.
Table 1: Internal validation RMSD value of 3G0B protein
| Scoring function | Moldock optime (Å) | Moldock SE (Å) | Iterated simplex (Å) |
| Moldock Score | 0.584487 | 0.599842 | 1.96978 |
| Moldock Score (Grid) | 0.816957 | 0.744002 | 0.829497 |
| Plant Score | 0.436046 | 0.490639 | 1.12917 |
| Plant Score (Grid) | 0.81555 | 0.383922 | 1.10598 |
Note: RMSD – Root mean square deviation; SE – Simplex evolution
Table 2: Internal validation RMSD value of 5Y7K protein
| Scoring function | Moldock optime (Å) | Moldock SE (Å) | Iterated simplex (Å) |
| Moldock Score | 6.38688 | 1.64357 | 1.48793 |
| Moldock Score (Grid) | 6.88049 | 3.27693 | 5.90506 |
| Plant Score | 6.80853 | 2.24238 | 2.86813 |
| Plant Score (Grid) | 5.05709 | 2.54698 | 2.32431 |
Note: RMSD-Root mean square deviation; SE-Simplex evolution
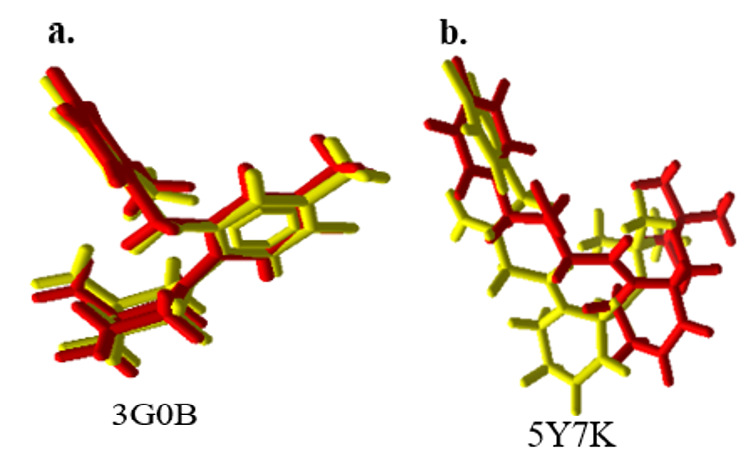
Fig. 1: Overlay of crystallographic native ligand/before docking (yellow) and redocked native ligand/after docking (red). a: 3G0B; b: 5Y7K
Molecular docking analysis
The results of the molecular docking studies presented in table 3 indicate that four compounds; 3,4-dicaffeoylquinic acid, nystose, 1,3-O-dicaffeoylquinic acid, and 3,5-dicaffeoylquinic acid-exhibit significantly lower rerank scores, ranging from-99.168 to-118.13 Kcal/mol. These scores are notably lower than that of sitagliptin (-91.9833 Kcal/mol), which serves as the positive control in this study. The rerank score serves as a pivotal parameter in molecular docking analyses, particularly for assessing the binding affinities of ligands to their respective target proteins. A lower rerank score typically signifies a more robust interaction between the ligand and the target, indicating an enhanced potential efficacy of the compound as a viable drug candidat [15]. Based on these findings, the four compounds demonstrate considerable potential as DPP-4 inhibitors, suggesting their viability for further investigation in the context of therapeutic applications for diabetes management. However, while the strong binding affinity indicated by the lower rerank scores suggests a promising interaction, it is imperative to validate these compounds through molecular dynamics simulations. Such simulations will provide insights into the stability and strength of the binding complexes formed between the compounds and their respective receptors. By assessing the dynamic behaviour of these complexes, we can gain a deeper understanding of the interactions at play, thereby confirming their potential as effective DPP-4 inhibitors. This step is crucial for elucidating the efficacy and reliability of the compounds in a biological context, ensuring that their in silico predictions translate into viable therapeutic candidates.
Table 3: Rerank score of docking results with molegro virtual docker
| S. No. | Plants | Compounds | Affinity-based on rerank score (Kcal/mol) |
| 1. | Smallanthus sonchifolius | 3,4-dicaffeoylquinic acid | -118.13 |
| 2. | Smallanthus sonchifolius | Nystose | -111.213 |
| 3. | Comparator Ligand | Alogliptin | -105.306 |
| 4. | Smallanthus sonchifolius | 1,3-O-dicaffeoylquinic acid | -103.511 |
| 5. | Smallanthus sonchifolius | 3,5-dicaffeoylquinic acid | -99.168 |
| 6. | Comparator Ligand | Sitagliptin | -91.9833 |
Visualization of the best docking results
The visualization of the Alogliptin complex as the reference ligand with protein 3G0B, as shown in fig. 2 and detailed in table 4, demonstrates that it interacts with the receptor's active site area through hydrogen bonding with amino acid residues Glu206, Glu205, Tyr547, Tyr631, and Ser630. Glu205 and Glu206 play essential roles in the inhibitory activity of DPP-4 inhibitors [16]. These amino acid residues are instrumental in establishing salt bridges with the positively charged portions of the inhibitors, thereby increasing both binding affinity and specificity. Furthermore, Glu205 and Glu206 help stabilize the ligand within the active site through electrostatic interactions, which aids in effectively blocking the enzyme's activity. The above residues make the interaction of the inhibitor with the enzyme rather stable, which is an essential factor for the intense action of DPP-4 inhibition. Hydrogen bonding defines the success of docking results and crucially affects the interactions during the binding of ligands to proteins. This bond is created by a hydrogen atom that is covalently bonded on the one hand to an electronegative atom, most often oxygen or nitrogen, and on the other, to another electronegative atom. This contact increases the specificity and stability of the ligand-protein complex. Notably, among the top four compounds identified in the docking results, Nystote demonstrates the highest number of hydrogen bonds, indicating a robust interaction profile and a structural similarity to the standard ligand. This suggests that Nystote may exhibit favorable binding characteristics, warranting further investigation into its potential as a lead compound in drug development.
Alogliptin |
Sitagliptin |
3,4-Dicaffeoylquinic acid |
Nystose |
1,3-O-dicaffeoylquinic acid |
3,5-dicaffeoylquinic acid |
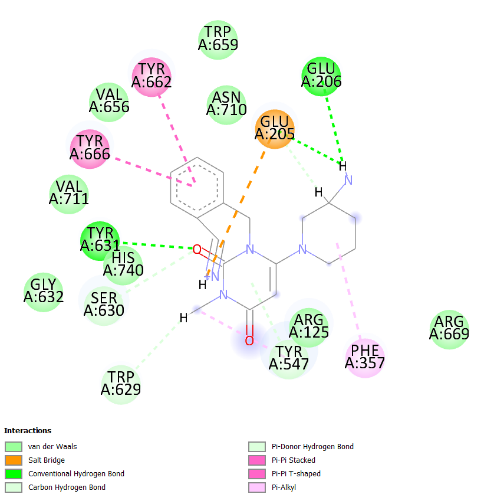
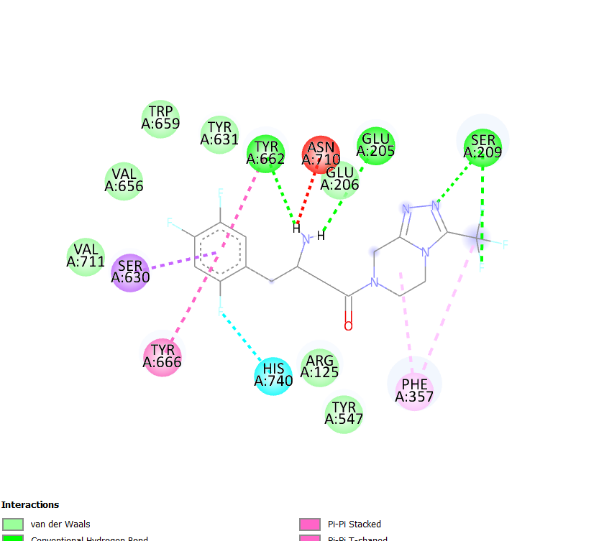
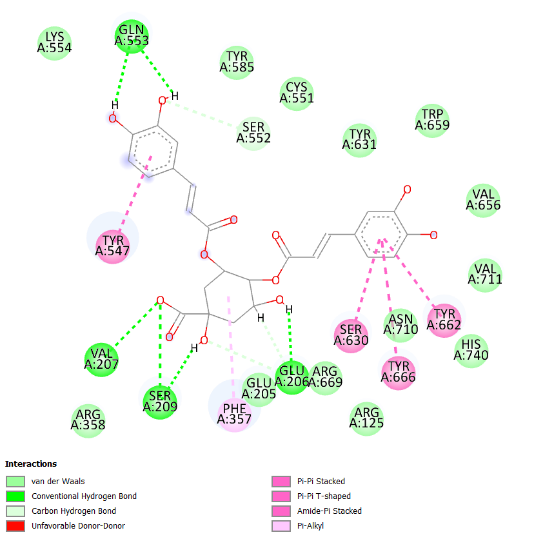
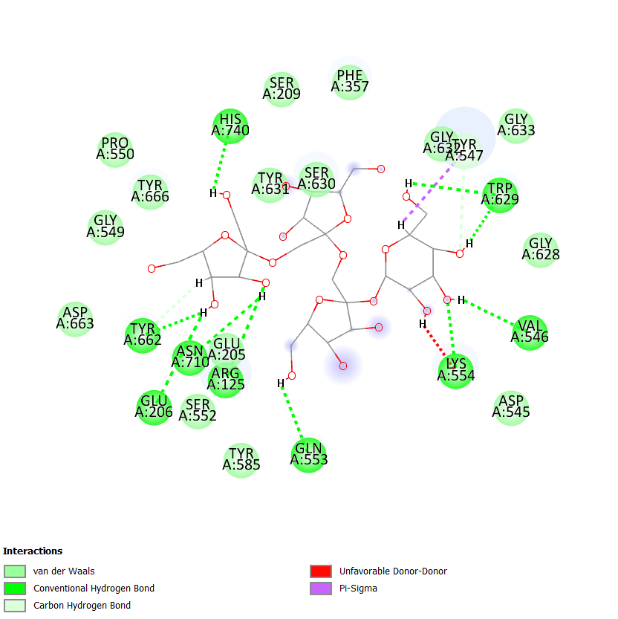
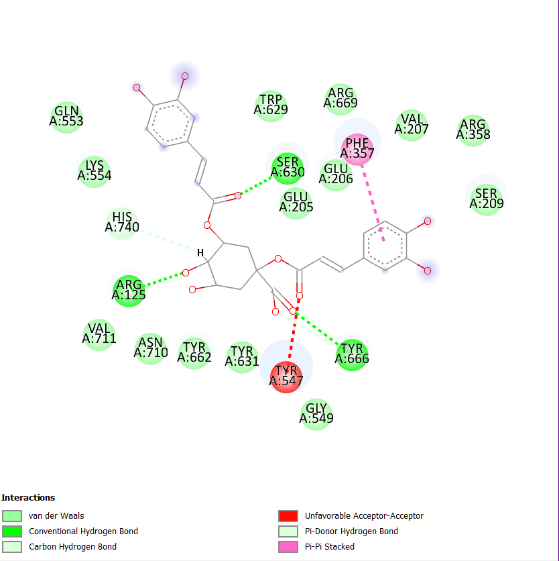
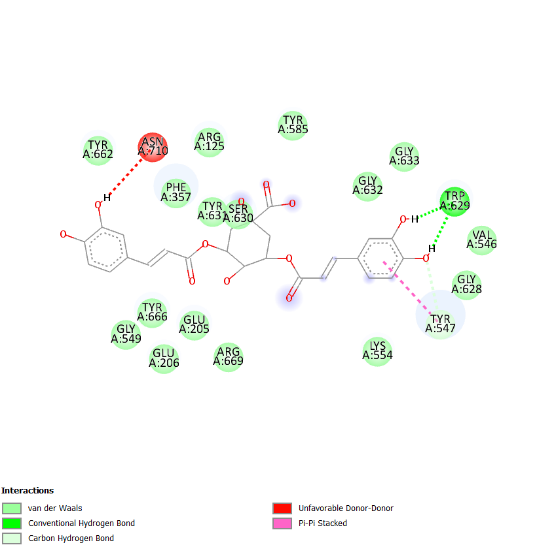
Fig. 2: Visualization of hydrogen bond interactions of compound amino acid residue
Table 4: Ligand complex amino acid residues
| No. | Compounds | Hydrogen bond amino acid | Non-hydrogen bond amino acid |
| 1. | Alogliptin | Glu206 (2.47 Å), Glu205 (2.82 Å), Tyr631 (2.19 Å), Tyr547 (2.45 Å), Ser630 (2.56 Å), Trp629 (2.80 Å) | Tyr662, Tyr666, Phe357, Trp659, Val656, Val711, His740, Gly632, Arg669 |
| 2. | Sitagliptin | Tyr547 (2.08 Å), Tyr662 (1.84 Å), Ser209 (2.21 Å), Glu205 (2.36 Å) | Glu206, Trp659, Val656, Val711, Ser630, Tyr666, His740, Arg125, Phe357 |
| 3. | 3,4-Dicaffeoylquinic Acid | Gln553 (2.26 Å), Glu206 (1.98 Å), Val207 (3.06 Å), Ser209 (2.92 Å) | Lys554, Tyr585, Cys551, Tyr631, Trp659, Val656, Val711, His740, Ser630, Tyr662, Tyr666, Asn710, His740, Arg125, Arg669, Arg125, Glu205, Phe357, Arg358, Tyr547, Lys554 |
| 4. | Nystose | His740 (2.36 Å), Trp629 (2.07 Å), Val546 (2.56 Å), Tyr662 (1.54 Å), Asn710 (2.77 Å), Glu206 (2.71 Å), Arg125 (2.26 Å), Gln553 (2.58 Å), Lys554 (2.17 Å), Tyr547 (2.53 Å) | Pro550, Tyr666, Gly549, Asp663, Glu205, Ser552, Tyr585, Asp545, Gly628, Gly633, Gly632, Phe357, Ser209 |
| 5. | Isochlorogenic acid C | His740 (2.15 Å), Tyr662 (2.68 Å), Arg669 (1.77 Å), Glu206 (1.96 Å), Ser209 (2.31 Å) | His126, Arg125, Arg358, Val207, Phe357, Glu205, Tyr631, Trp659, Val656, Val711, Asn710, Ser630, Tyr547 |
| 5. | 1,3-O-Dicaffeoylquinic acid | Ser209 (2.06 Å), Glu206 (1.47 Å), Arg669 (2.31 Å), His740 (2.62 Å) | Gln553, Lys554, Val711, Asn710, Tyr662, Tyr631, Gly549, Tyr547, Ser209, Arg358, Val207, Arg669, Trp629, Glu205 |
| 6. | 3,5-dicaffeoylquinic acid | Trp629 (1.81 Å),Tyr547 (2.85 Å) | Tyr662, Asn710, Arg125, Phe357, Tyr631, Ser630, Tyr585, Gly632, Gly633, Val546, Gly628, Lys554, Arg669, Glu205, Glu206, Tyr666, Gly549 |
ADMET analysis
Percentage of human intestinal absorption (HIA) determines the probability that an administered compound will be absorbed, prediction its HIA profile. So, absorption under HIA value is divided with respect to percentage of absorbed drug i. e. Poor; 0-20%, moderate mean; upto that level it can be considered as recognized and useful,effective, last one Good 70-100% reflect good absorptions [17]. Table 05 the %HIA values of three tested compounds by the current study therefore, 3,4-dicaffeoylquinic acid and the related compounds; wich is predecessors to produce by human metabolism (1,3-O-dicaffeoyl quinic-acid) and a subtype of this group that are not conceivable consequences present estimated moderate absorption rates in humans. However, data in table 5 evidenced Nystose with poor permeability and HIA of 0%. Enhanced solubility and permeability of Nystose could potent for oral absorption improvement using other modifications in the molecular structure or formulations.
The blood-brain barrier (BBB) is a diffusion-and charge-based selective-semipermeable membrane that separates the circulating blood from the brain interstitium whereby the properties of BBB allow specialized function(s) to be performed by human cerebral microvessels including regulation control at metric levels on passing certain molecules and ions while others are restricted between plasma in vessel wall compartments [18]. In particular, compounds with a positive Log BB (>0.3) generally pass the BBB very well and those with an unfavorableLogBB value (<-1), will distribute poorly into the brain. In this study, the BBB permeability values for the test compounds ranged from-2.099 to-1.983. All four of the best test compounds indicated an inability to penetrate the blood-brain barrier. However, since the DPP-4 inhibitor diabetes medications interact mainly with insulin (the hormone secreted by your pancreas), and hence control blood sugar through the endocrine system (rather than nervous system), they do not need to cross brain's protect. Considering that both the test compounds have to work in periphery, their inability for BBB permeability does not matter, so we are let less bothered about predicted outcome-related effects on BBB.
CYP3A4 and CYP2D6 isoenzymes represent>50% of the cytochrome P450 (Cyp) associated drug metabolism. CYP3A4, found at the highest levels in humans and responsible for metabolism of more than 30% of drugs alone, CYP2D6 (accounting for nearly one-fifth metabolic processes) Among our metabolic predictions, 3 substrates of CYP3A4 were identified: i. e. 3,4-dicaffeoylquinic acid, 1,3-O-dicaffeoylquinic acids and 3,5-dicaffeoylquinic acid. This result shows that these compounds are metabolized by CYP450 enzymes. In contrast, nystose is a non-substrate of CYP3A4, indicating that it can not be metabolized by this pathway. Thus, nystose may be have a different pharmacological profile in comparison with remaining compounds. Furthermore, none of these compounds inhibited CYP2D6 or 3A4 at the concentrations tested indicating that they are unlikely to affect the pharmacokinetics of co-administered drugs. The predictive results suggested that three compounds (75%) could bind to CYP3A4 in drug metabolism, implying possible pharmacological implications.
The prediction of a compound's excretion can be evaluated by assessing the total clearance rate, which encompasses both liver clearance (metabolism in the liver and bile) and renal clearance (excretion via the kidneys). Total clearance is categorized as low when the clearance value is<2 ml/min/kg, moderate when it ranges from 2 to 15 ml/min/kg, high when it falls between15 and 20 ml/min/kg, and very high when it exceeds 20 ml/min/kg. In the present study, the total clearance values of the four best test compounds ranged from-0.056 to 1.772 ml/min/kg. Notably, the Nystose compound exhibited the highest total clearance value, indicating that it is likely to be eliminated from the body more rapidly than the other compounds, which have lower total clearance values. This suggests that Nystose may have a shorter duration of action and could require more frequent dosing in therapeutic applications, emphasizing the importance of considering its pharmacokinetic profile in clinical settings.
The LD50 value is classified into six groups of super toxic (≤5 mg/kg), highly toxic (5-50 mg/kg), moderately toxic (500–2000 mg/kg), lowly other type classification namely differentiated stimuli and noxious chemicals relevance to the non-target organism, reference pesticide comparison or etc. As indicated in table 5, the estimated LD50 values for all four compounds are between 1325.6 and 2258.73 mg/kg meaning that these substances belong to non-toxic category.
Analysis of molecular dynamics results
Molecular dynamics is a computational simulation technique employed in the theoretical investigation of biological molecules, including proteins and nucleic acids, to examine the physical motion of their constituent atoms and molecules. This simulation method is also utilized to assess the stability of interactions between proteins and ligands under conditions that closely replicate the physiological environment of the human body over a specified duration. In this study, molecular dynamics simulations were conducted on the test ligands exhibiting the most negative re-rank scores. The selected test ligands for simulation included 3,4-dicaffeoylquinic acid, nystose, 1,3-O-dicaffeoylquinic acid, and 3,5-dicaffeoylquinic acid.
RSMD
An increase in the root mean square deviation (RMSD) value suggests that the protein structure begins to exhibit flexibility or opens up, allowing the ligand to explore potential binding sites or coordinates within the protein. Conversely, a stable RMSD value signifies that the protein has achieved a stable conformation in its interaction with the ligand, indicating that the optimal binding conformation has been reached. This stability is crucial for assessing the reliability of ligand-protein interactions in molecular dynamics simulations (fig. 3).
Table 5: Compound profile absorption, distribution, metabolism, excretion, and toxicity
| S. No. | Compounds test | Absorption | Distribution | Metabolism | Excretion | Toxicity | |
| Intestinal absorption (%) | BBB Permeability | Inhibitor CYP2D6 | Substrate CYP2D6 | Substrate CYP3A4 | Inhibitor CYP3A4 |
||
| 1 | 3,4-dicaffeoylquinic acid | 29.037 | -2.08 | No | No | Yes | No |
| 2 | Nystose | 0 | -2.099 | No | No | No | No |
| 3 | 1,3-O-dicaffeoylquinic acid | 30.305 | -1.983 | No | No | Yes | No |
| 4 | 3,5-dicaffeoylquinic acid | 44.225 | -2.069 | No | No | Yes | No |
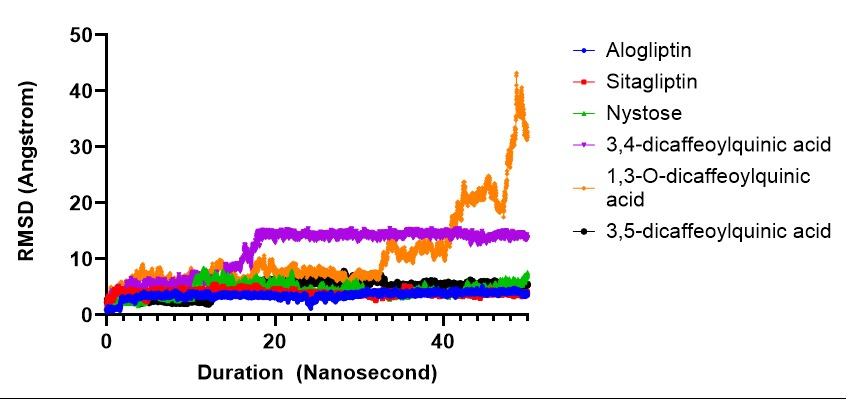
Fig. 3: Ligand movement RMSD after superposing on the receptor DPP-4
The RMSD (fig. 3) analyses of alogliptin and sitagliptin, utilized as positive controls in the molecular dynamics simulations, indicated stable conformational dynamics over a 50 ns duration. The average RMSD values for alogliptin and sitagliptin were 3.539 Å and 3.931 Å, respectively. This is indicative that the two compounds maintain their structural stability over the course of all MD simulations and, therefore could be potentially used as therapeutic agents in clinical settings. The behavior is more interesting for 3,4-dicaffeoylquinic acid and 1,3-O-dicaffeoylquininc where they show a significant conformational change in the protein-ligand complex. In both cases, the RMSD of values showed more than 10 Å, indicating large fluctuations in the system that may cause disturbance to structural integrity or lessen ligands ability to remain a stable pose for binding. This observed instability indicates that these compounds may not interact optimally with their target proteins, potentially undermining their pharmacological efficacy. The RMSD analysis of 3,5-dicaffeoylquinic acid reveals overall unstable conformational dynamics throughout the 50 ns simulation period. Conformational changes tend to increase from 15 ns to 50 ns, with RMSD values exceeding 4 Å and failing to reach a stable conformation. This persistent instability suggests that 3,5-caffeoylquinic acid may struggle to maintain a consistent binding conformation within the protein-ligand complex, which could adversely affect its interaction with the target protein and its overall pharmacological potential.
The RMSD analysis of the nystose test compound revealed overall stable conformational dynamics throughout the 50 ns simulation period. Fluctuations were observed between 10 and 20 ns, with the RMSD value rising to approximately 6-7 Å. However, from 20 to 50 ns, the RMSD value decreased, indicating a return to a more consistent conformation. The average RMSD value for nystose was calculated to be 4.567 Å. These findings suggest that nystose maintains its structural integrity despite experiencing transient conformational fluctuations. Although the in silico results indicate stability, further investigation through in vitro or in vivo studies is necessary to validate these findings and to gain deeper insights into the biological implications of nystose.
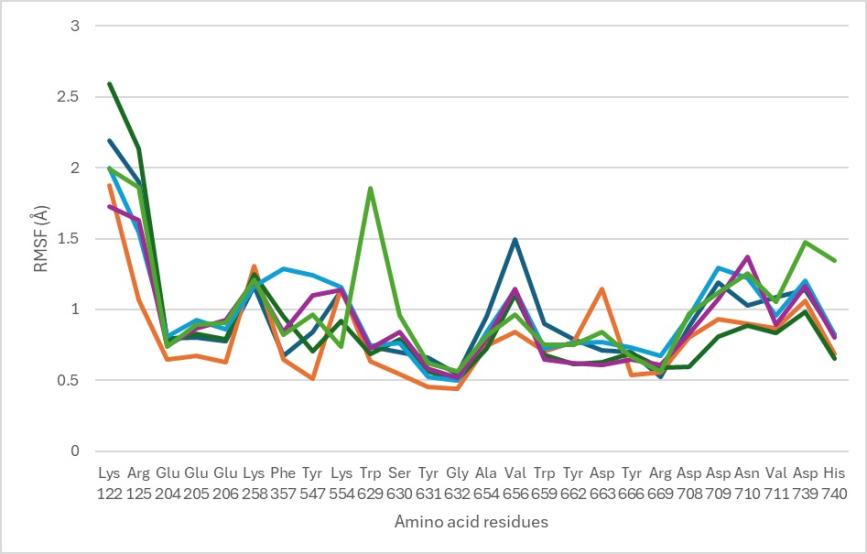
Fig. 4: The root mean square fluctuation (RMSF) value of the DPP4 residues binding site
RMSF
The RMSF analysis was performed to assess the flexibility of amino acid residues during the molecular dynamics simulation. Fig. 4 illustrates the fluctuations between the ligand and the amino acid residues within the enzyme. The critical amino acid residues involved in the attachment process with the test ligand during molecular dynamics simulations include Glu205, Glu206, Ser209, Tyr547, Tyr631, Tyr662, and Ser630. Based on these findings, the four compounds did not cause significant (<3 Å) fluctuations on the amino acid residues at their respective binding sites, indicating a stable interaction between the ligand and the enzyme during the simulation.
Binding energy
The binding energy calculations were performed using YASARA-based molecular dynamics simulations to assess the thermodynamic stability and affinity between the ligand and the target protein. The analysis aimed to elucidate the key molecular interactions contributing to complex formation and stability. The average binding energy throughout the simulation was calculated to be X kJ/mol, indicative of a strong and energetically favorable binding interaction. In Yasara, a lower (more negative) binding energy typically suggests a weaker interaction between the ligand and the target protein, indicating that more energy is required to sustain the complex, thereby reflecting a reduced binding affinity. As the binding energy increases, the stability of the complex may diminish, implying that the ligand binds less effectively to the target. Conversely, A higher (less negative) binding energy denotes a more stable and favorable interaction, indicative of a stronger binding affinity and a thermodynamically favorable binding process. The binding energy analysis (fig. 5) reveals that the four compounds exhibit lower average binding energy values (ranging from 243.905 kJ/mol to 314.61 kJ/mol) compared to the positive control, indicating that their stability is lower than that of the positive control. Based on this observation, the dosage of the test compounds needs to be increased to achieve biological activity comparable to that of the positive control.
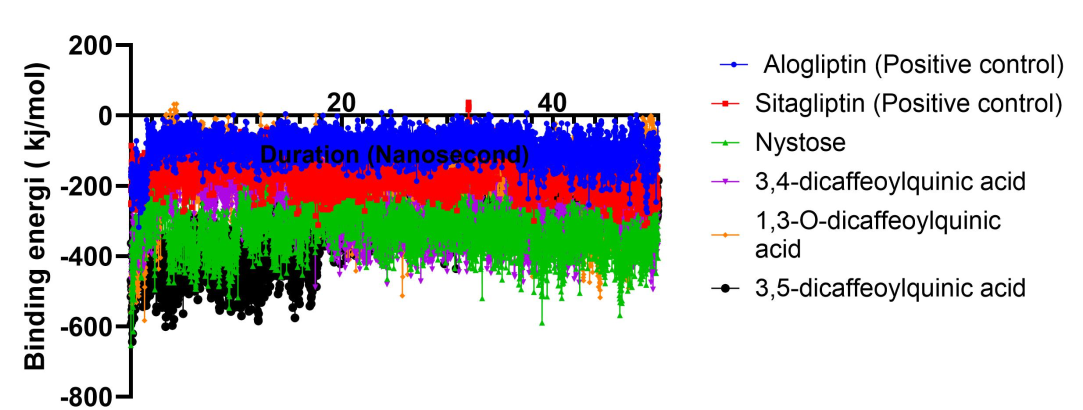
Fig. 5: The free energy of binding value ligand-DPP4
DISCUSSION
Internal validation via redocking is a critical parameter for assessing the accuracy and robustness of docking algorithms. In this process, the crystallized protein-ligand complex is separated from its ligand without undergoing energy minimization, after which the ligand is re-docked into its original receptor binding site [14]. This method evaluates whether a docking approach can reliably reproduce experimental binding poses, as determined by structural techniques such as X-ray crystallography. The primary outcome of this validation is the root-mean-square deviation (RMSD) between the original and redocked poses, with RMSD values ≤ 2 Å generally considered acceptable. Lower RMSD values indicate a greater similarity between the predicted and experimental ligand positions. In the case of the 5Y7K protein (table 2), the native ligand from the crystallographic data and the redocking results exhibited minimal overlap, with RMSD values exceeding 2 Å across all docking protocol combinations. This deviation reflects a significant difference between the crystallographic ligand orientation and the docked pose, as visually confirmed by the non-overlapping positions. However, in the case of the 3G0B protein (table 1), the native ligand from the crystallographic data and the redocking results demonstrated significant overlap, with RMSD values below 2 Å (approaching 0 Å) across all docking protocol combinations. This indicates that the docking protocol validation using the 30B protein successfully positioned the native ligand at the correct binding site. Achieving accurate placement of the ligand is crucial, as it enhances the reliability of the docking model for predicting binding interactions, thus improving the overall accuracy of virtual screening and drug discovery efforts. These findings are consistent with those reported by Yogini et al. (2024), where a similar RMSD threshold was used to validate docking protocols for protein 4C15, resulting in accurate ligand placement and enhanced predictive performance [19]. On the other hand, research conducted by Sachdeo et al. (2024) demonstrated that docking models with RMSD values greater than 2 Å tend to exhibit reduced predictive accuracy, often leading to incorrect binding poses and diminished utility in drug discovery [20]. By achieving an RMSD close to 0 Å, the docking validation in this study ensures a higher degree of reliability, comparable to the most accurate models reported in the literature.
Based on table 3, 4 test compounds have better affinity than the comparator compounds, namely 3 compounds that are better than native ligand and 2 compounds that are better than sitagliptin. This result can be stated based on the comparison of the re-rank score value, which is more negative than the comparator compound. The compounds are 3,4-dicaffeoylquinic acid, nystose, 1,3-O-dicaffeoylquinic acid, and 3,5-dicaffeoylquinic acid. Rerank Score describes the bonding of a ligand with its receptor. The value of the rerank score reflects the bonding energy required to form a bond between the ligand and its receptor, thereby predicting its activity. The lower the rerank score, the more stable the bond between the ligand and the receptor, and the higher the activity of the compound with the lowest rerank score value is predicted. On this basis, it is predicted that the 4 compounds have good activity [21]. Consistent with studies by Imai et al. (2020), who also showed the antidiabetic action of caffeoylquinic acid derivatives to be related both to DPP-4 inhibition and glucose tolerance enhancement [22]. Moreover, studies by Alam et al. (2022) emphasize that nystose, a fructooligosaccharide, has shown antidiabetic effects by enhancing gut microbiota composition and improving insulin sensitivity, which complements its predicted DPP-4 inhibition profile found here [23]. Therefore, the strong binding affinities and lower reranking scores of these four compounds suggest not only their robust DPP-4 inhibitory activity but also their potential to contribute to improved glycemic control, making them promising candidates for further investigation in the treatment of type 2 diabetes.
The visualization process of the molecular docking results aims to observe and identify the similarities between amino acid residues and the bonds between ligands and receptors. Ligands in test compounds that interact with amino acid residues and form bonds similar to ligands in comparator compounds show similar activity because they involve the same type of interaction. The amino acid residues Glu205, and partly the two adjacent "clamping" glutamate, which anchor the N-terminal atom at P2 position of GLP-1 (glucagon-like peptide 1) incretin hormone agonist/GIP substrate to DPP-4 protein would be very pocetent is those site hhave stron interaction with enzyme conversed on binding siten if that inhibitor able tightly bind especially between Ser630 to His740 and Ser209 resulting in prevention compositional interchange so as table. Because of those results, we conducted the docking analysis to determine other amino acid residues that were crucial for inhibition DPP-4 enzyme action in addition Glu205 and 206. Based on the results of the 2D visualization of the ligand-receptor that has been done, several test compounds have interactions on the same amino acid residues listed in the table. This indicates the same mechanism of action between the test compound and the comparison compound. In addition to Glu205 and Glu206, the docking analysis identified other important amino acid residues, including Ser209, Ser630, Tyr547, and Tyr662, which are also significant for the inhibition of DPP-4 enzyme activit. The 2D visualization results of the ligand-receptor interactions reveal that several test compounds interact with the same amino acid residues identified in the table. This observation indicates a shared mechanism of action between the test compounds and the comparator compound, consistent with findings from Choi et al. (2019), who reported similar binding interactions for other DPP-4 inhibitors [24]. Such parallels underscore the importance of targeting these amino acid residues for the development of effective antidiabetic therapies.
Root mean Square Deviation (RMSD) is a valuable metric for evaluating molecular stability and observing conformational changes over time during molecular dynamics simulations. It measures how structures deviate from their initial conformation, providing insight into the stability of ligand binding. In this study, only Nystose demonstrated an RMSD consistently below 3 Å, reflecting a highly stable interaction with the DPP-4 enzyme. This minimal deviation suggests that Nystose maintains a firm binding within the active site, indicative of a stable inhibitory potential. Comparatively, previous studies have highlighted the importance of low RMSD values in identifying strong DPP-4 inhibitors. Research conducted by Du et al. l (2016) showed RMSD values below 3 Å tended to exhibit enhanced binding stability and, consequently, higher inhibitory efficacy [25]. The results for Nystose align with these findings, reinforcing that maintaining a stable RMSD profile is critical for achieving optimal binding affinity and stability in DPP-4 inhibitors derived from natural compounds. This stability suggests that Nystose, and potentially other compounds with low RMSD, may serve as effective DPP-4 inhibitors. This further supports the potential of Smallanthus sonchifolius derivatives in developing plant-based antidiabetic treatments, highlighting their relevance for future in vitro studies and drug development.
The RMSF analysis demonstrated that critical amino acid residues at the DPP-4 binding site, specifically Glu205, Glu206, Ser209, Tyr547, Tyr631, Tyr662, and Ser630, exhibited minimal fluctuations, with average RMSF values remaining consistently below 3Å. This low fluctuation indicates high structural stability of these residues in their interactions with the ligand, essential for strong and sustained binding during inhibition. Such findings align with previous studies where stable binding site interactions, particularly with Glu and Tyr residues, were shown to play a pivotal role in maintaining inhibitory effectiveness. For instance, studies by Fatimah et al. l (2024) also reported low RMSF values for similar critical residues, correlating stable binding with enhanced inhibitory activity [26]. These results support the hypothesis that maintaining low flexibility within these critical residues enhances the ligand’s efficacy in stabilizing the DPP-4 complex, potentially contributing to a more efficient inhibition mechanism compared to ligands that induce higher flexibility in the binding pocket.
The four test compounds demonstrated lower binding energy values compared to the positive control, indicating a potentially lower interaction within the binding site. In molecular dynamics simulations with yasara, binding energy is defined as the difference in free energy between the bound state and the fully unbound state (where binding pairs are at an infinite distance). According to this definition, a more positive binding energy suggests stronger binding affinity. Comparatively, previous studies have shown that stronger binding energies correlate with improved inhibition efficacy, as noted by Odhar et al. (2020), underscoring that compounds with more negative binding energy, such as those observed here, may serve as promising candidates for DPP-4 inhibition [27, 28].
CONCLUSION
Molecular docking investigations of identified small anthussonchifolius compounds to the DPP-4 3G0B protein included suggesting a high binding efficacy for four potential candidates. The results of docking studies indicate that these compounds interact with the main amino acids residues in a very similar mode as seen for an already known DPP-4 inhibitor, sitagliptin (active compound), and glycyl-L-proline which is a native ligand of this enzyme. The inhibitory capability and binding stability of these compounds were then confirmed by further in-depth molecular dynamics simulations. Nystose appeared to be the most stable in terms of RMSD among tested compounds and indicated its best inhibitory activity. Moreover, the RMSF analysis corroborated these results as they showed stable interactions with key amino acid residues of the DPP-4 enzyme, and their fluctuations remained less extensive suggesting that it tightly binds to the active site for all tested primary compounds. in terms of binding energy, 1,3-O-Dicaffeoylquinic acid showed the most favorable energy, followed by 3,4-Dicaffeoylquinic acid, 3,5-Dicaffeoylquinic acid, and finally, Nystose. While Nystose demonstrated significant stability, the high binding affinity of caffeoylquinic acid derivatives underscores their potential as potent DPP-4 inhibitors. These findings suggest that natural DPP-4 inhibitors from Smallanthus sonchifolius could inspire future in vitro studies and may hold promise in the development of plant-derived therapies for diabetes management.
FUNDING
This research was funded by [Insert Funding Source or Organization], which provided financial support for the data collection, analysis, and publication process.
AUTHORS CONTRIBUTIONS
Novi Yantih: Conceptualization, Methodology, Writing-review, Supervision. Zuhelmi Aziz: Investigation, Writing-review, Formal analysis. Esti Mumpuni: Methodology, Writing-review, Resources. Nurul Widayanti: Data curation, Wiriting-review, Visualization. Andri Prasetiyo: Conceptualization, Writing-original draft, Review and editing, Visualization.
CONFLICT OF INTERESTS
Declared none
REFERENCES
International Diabetes Federation International Diabetes Federation. IDF diabetes atlas. 10th ed. Boyko EJ, Magliano DJ, Karuranga S, Piemonte L, Riley P, Saeedi P. editors; 2021.
Salvo F, Moore N, Arnaud M, Robinson P, Raschi E, DE Ponti F. Addition of dipeptidyl peptidase-4 inhibitors to sulphonylureas and risk of hypoglycaemia: systematic review and meta-analysis. BMJ. 2016;353:2231. doi: 10.1136/bmj.i2231, PMID 27142267.
D Andrea E, Wexler DJ, Kim SC, Paik JM, Alt E, Patorno E. Comparing the effectiveness and safety of SGLT2 inhibitors vs DPP-4 inhibitors in patients with type 2 diabetes and varying baseline HbA1c levels. JAMA Intern Med. 2023;183(3):242-54. doi: 10.1001/jamainternmed.2022.6664, PMID 36745425.
Natale P, Palmer SC, Tunnicliffe DJ, Toyama T, Strippoli GF. Thiazolidinediones for people with chronic kidney disease and diabetes. Cochrane Database Syst Rev. 2023 Sep 7;2023(9):CD015907.
Chandra V, Yadav P, Raghuvanshi V, Pandey P. Diabetes and ethnomedicine a comprehensive review of scientific literature on traditional medical practices. J Pharm Res. 2023;8(2).
Riyanti S, Suganda AG, Sukandar EY. Dipeptidyl peptidase IV inhibitory activity of some Indonesian medicinal plants. Asian J Pharm Clin Res. 2016;9(2):375-7.
Baroni S, Suzuki Kemmelmeier F, Caparroz Assef SM, Cuman RK, Bersani Amado CA. Effect of crude extracts of leaves of smallanthus sonchifolius (yacon) on glycemia in diabetic rats. Rev Bras Cienc Farm. 2008;44(3):521-30. doi: 10.1590/S1516-93322008000300024.
Hachkova H, Nagalievska M, Soliljak Z, Kanyuka O, Kucharska AZ, Sokol Letowska A. Medicinal plants Galega officinalis l. and yacon leaves as potential sources of antidiabetic drugs. Antioxidants (Basel). 2021;10(9):1362. doi: 10.3390/antiox10091362, PMID 34572994.
Brogi S, Ramalho TC, Kuca K, Medina Franco JL, Valko M. Editorial: in silico methods for drug design and discovery. Front Chem. 2020 Aug 7;8:612. doi: 10.3389/fchem.2020.00612, PMID 32850641.
Prasetiyo A, Mumpuni E, Luthfiana D, Herowati R, Putra GS. In silico discovery of potential sodium glucose cotransporter-2 (SGLT-2) inhibitors from Smallanthus sonchifolius (Poepp.) H. Rob. Via molecular docking and molecular dynamics simulation approach. Journal of Pharmacy & Pharmacognosy Research 2025;13(3):716-28. doi: 10.56499/jppres24.2104_13.3.716.
Komura H, Watanabe R, Mizuguchi K. The trends and future prospective of in silico models from the viewpoint of ADME evaluation in drug discovery. Pharmaceutics. 2023;15(11):2619. doi: 10.3390/pharmaceutics15112619, PMID 38004597.
Bitencourt Ferreira G, DE Azevedo WF. Molegro virtual docker for docking. Methods Mol Biol. 2019;2053:149-67. doi: 10.1007/978-1-4939-9752-7_10, PMID 31452104.
Torres PH, Sodero AC, Jofily P, Silva JR FP. Key topics in molecular docking for drug design. Int J Mol Sci. 2019;20(18):4574. doi: 10.3390/ijms20184574, PMID 31540192.
Prasetiyo A, Kumala S, Mumpuni E, Tjandrawinata RR. Validation of structural based virtual screening protocols with the PDB Code 3G0B and prediction of the activity of Tinospora crispa compounds as inhibitors of dipeptidyl peptidase IV. Res Results Pharmacol. 2022;8(1):95-102. doi: 10.3897/rrpharmacology.8.76237.
Edache EI, Uzairu A, Shallangwa GA, Mamza PA. Virtual screening pharmacokinetics and molecular dynamics simulations studies to identify potent approved drugs for chlamydia trachomatis treatment. Futur J Pharm Sci. 2021;7(1). doi: 10.1186/s43094-021-00367-4.
Farkhani A, Sauriasari R, Yanuar A. In silico approach for screening of the Indonesian medicinal plants database to discover potential dipeptidyl peptidase-4 inhibitors. Int J Appl Pharm. 2020;12(1):60-8.
Bitew M, Desalegn T, Demissie TB, Belayneh A, Endale M, Eswaramoorthy R. Pharmacokinetics and drug likeness of antidiabetic flavonoids: molecular docking and DFT study. Plos One. 2021 Dec;16(12):e0260853. doi: 10.1371/journal.pone.0260853, PMID 34890431.
WU D, Chen Q, Chen X, Han F, Chen Z, Wang Y. The blood brain barrier: structure regulation and drug delivery. Signal Transduct Target Ther. 2023;8(1):217. doi: 10.1038/s41392-023-01481-w, PMID 37231000.
Yogini CS, Kumar CS, Anuradha CM, Chitturi CH. Computational assessment of Undaria pinnatifida and Moringa oleifera compounds as anti-obesity agents. Int J App Pharm. 2024;16(5):309-26. doi: 10.22159/ijap.2024v16i5.50867.
Maeda K, Das D, Kobayakawa T, Tamamura H, Takeuchi H. Discovery and development of anti-HIV therapeutic agents: progress towards improved HIV medication. Curr Top Med Chem. 2019;19(18):1621-49. doi: 10.2174/1568026619666190712204603, PMID 31424371.
Prasetiyo A, Mumpuni E, Luthfiana D, Herowati R, Putra GS. In silico discovery of potential sodium glucose cotransporter-2 (SGLT-2) inhibitors from smallanthus sonchifolius (Poepp.) H. Rob. Via molecular docking and molecular dynamics simulation approach. Journal of Pharmacy & Pharmacognosy Research 2025;13(3):716-28.
Imai M, Yamane T, Kozuka M, Takenaka S, Sakamoto T, Ishida T. Caffeoylquinic acids from aronia juice inhibit both dipeptidyl peptidase IV and α-glucosidase activities. LWT. 2020;129:109544. doi: 10.1016/j.lwt.2020.109544.
Alam S, Sarker MM, Sultana TN, Chowdhury MN, Rashid MA, Chaity NI. Antidiabetic phytochemicals from medicinal plants: prospective candidates for new drug discovery and development. Front Endocrinol (Lausanne). 2022 Dec;13:800714. doi: 10.3389/fendo.2022.800714, PMID 35282429.
Batool M, Ahmad B, Choi S. A structure based drug discovery paradigm. Int J Mol Sci. 2019;20(11):2783. doi: 10.3390/ijms20112783, PMID 31174387.
DU X, LI Y, Xia YL, AI SM, Liang J, Sang P. Insights into protein ligand interactions: mechanisms models and methods. Int J Mol Sci. 2016;17(2):144. doi: 10.3390/ijms17020144, PMID 26821017.
James JP, Srinivasa MG, Sindhu TJ, Jouhara BM, Revanasiddappa BC, C ZF. Invetigating multi target potential mucuna pruriens park dis insight from molecular docking MMGBSA pharmacophore modeling MD simulation and ADMET analysis. International Journal of Applied Pharmaceutics 2024;16(5):176-93. doi: 10.22159/ijap.2024v16i5.51474.
Odhar HA, Hashim AF, Ahjel SW, Humadi SS. Molecular docking and dynamics simulation analysis of the human FXIIa with compounds from the Mcule database. Bioinformation. 2023;19(2):160-6. doi: 10.6026/97320630019160, PMID 37814681.
Istyastono EP, Octa Riswanto FD. Molecular dynamics simulations of the caffeic acid interactions to dipeptidyl peptidase IV. Int J App Pharm. 2022;14(4):274-8. doi: 10.22159/ijap.2022v14i4.44631.