
Int J App Pharm, Vol 17, Issue 2, 2025, 259-267Original Article
LIQUISOLID COMPACT OF MECLIZINE HYDROCHLORIDE: DEVELOPMENT AND OPTIMIZATION USING FACTORIAL DESIGN
TULARAM BAROT1*, KARUNA NAGULA2, MOKSHA PATEL3, L. D. PATEL4
1Parul Institute of Pharmacy and Research, Faculty of Pharmacy, Parul University, Vadodara, India. 2Department of Pharmaceutics, College of Pharmaceutical Sciences, Dayananda Sagar University, Bengaluru, Karnataka, India. 3Department of Pharmaceutics, ROFEL, Shri G. M. Bilakhia College of Pharmacy, Vapi, Gujarat, India. 4Ex Director PG, Faculty of Pharmacy, Parul University, Vadodara, India
*Corresponding author: Tularam Barot; *Email: tularam.barot24711@paruluniversity.ac.in
Received: 07 Dec 2024, Revised and Accepted: 06 Feb 2025
ABSTRACT
Objective: The study focuses on improving the dissolution rate of meclizine hydrochloride by developing a liquisolid compact.
Methods: Meclizine hydrochloride is used to prevent motion sickness but has slow dissolution, requiring it to be taken an hour before travel. Various water-miscible solvents were examined to determine the drug's solubility, with propylene glycol showing the highest solubility. Avicel® PH 102 was chosen as the carrier, and Aerosil® 200 as the coating material. The formulation was optimized using Design Expert software and 32 factorial design was used to study the effects of factors: carrier and coating ratio(X1) and drug concentration in liquid medication (%Cd)(X2) on responses: %cumulative drug release at 20 min (Y1) and angle of repose (Y2).
Results: The optimized formulation was selected using the software, with a carrier and coating ratio of 24.89 and drug concentration in liquid at 10.14% w/w. The optimized liquisolid tablet was evaluated for post-compression parameters and dissolution study. The drug was found to be non-crystalline based on Differential Scanning Calorimetry (DSC) and X-ray diffraction (XRD) studies, and the stability study showed no significant degradation.
Conclusion: The findings indicate that liquisolid formulation can be a promising alternative to achieve rapid onset of action and dissolution enhancement for poorly water-soluble drugs such as meclizine hydrochloride.
Keywords: Meclizine hydrochloride, Liquisolid compact, Factorial design, Motion sickness
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i2.53361 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Meclizine hydrochloride is categorized as a Biopharmaceutical Classification System (BCS) class II drug due to its low solubility in water and high permeability. This medication is utilized as an antiemetic to prevent motion sickness, nausea, and vomiting and is typically administered one hour before traveling due to its slow dissolution property [1]. Meclizine hydrochloride is considered practically insoluble in water, with very low solubility [2-4]. Meclizine hydrochloride is available in the market in the form of tablet and capsule and they have a one-hour lag time in the onset of action because of its poor dissolution [5, 6]. Liquisolid compact is a promising formulation strategy for improving the dissolution rate of drugs with poor water solubility [7]. The rationale of current research is to develop a liquisolid dosage form in which the drug is pre-dissolved in a water-miscible vehicle to improve its wetting and dissolution characteristics. This technique involves dissolving the drug in a non-volatile, water-soluble solvent and then absorbing the resulting liquid mixture into a carrier material. To maintain good flow and compression properties, the liquid medication is added to the powder mixture in limited quantities, and the amount of liquid that can be added can be calculated using an equation [8]. As the carrier material is saturated with liquid medication, it leaves a thin layer of liquid on particle surface, which is instantly adsorbed by the coating material, which prevents the liquid medication from leaking out [9]. By employing the liquisolid compact technology to formulate meclizine hydrochloride, the dissolution rate can be improved, resulting in a faster onset of action. Liquisolid compacts are widely used for enhancing aqueous solubility, bioavailability, and reducing dissolution time due to an increase in surface area and a decrease in wetting angle [10, 11]. This technique can be scaled up to an industrial scale, enabling the manufacturing of sustained and controlled-release solid dosage forms. Appropriate concentration and selection of carrier and coating materials are critical to maintaining acceptable flow properties and compression characteristics of the final product [12, 13].
The main objective of the current research is to fasten the dissolution of the drug meclizine hydrochloride, which is approved as an over-the-counter (OTC) drug by United States Food and Drug Administration (USFDA) for motion sickness [14]. Motion sickness requires immediate action of the drug, which is difficult to achieve in the conventional dosage form of meclizine hydrochloride because of low solubility and poor wetting properties. By developing a liquisolid compact of meclizine hydrochloride, the drug is provided in pre-dissolved form in water miscible, non-volatile vehicle, which upon contact with Gastro-Intestinal (GI) fluids, immediately release the drug. Thus leading quick action of the drug [15, 16].
MATERIALS AND METHODS
Materials
Meclizine hydrochloride USP, received as a complimentary sample from Exemed Pharmaceuticals in Vapi, India, was used in the study. Different grades of Polyethylene Glycol (PEG 200, 400, 600), Glycerin, Sodium Starch Glycolate (SSG), Tween® 20, Aerosil® 200 (Batch Number 2FF0101), Tween® 80, Lactose Monohydrate, Avicel® PH 102 (Batch Number BCCH9027), Maize Starch, Propylene Glycol (PG), and Magnesium Stearate, were sourced from LobaChemie Pvt Ltd, Mumbai. Hydrochloric acid (HCl) and distilled water were obtained from Avantor Ltd., Mumbai, and Priya Chemicals, Vapi, respectively. All materials used were of laboratory grade.
Experimental part
Solubility study
The calibration curve of meclizine hydrochloride was developed in methanol as solvent using UV/Visible Spectrophotometer (Shimadzu UV-1800, Japan) analysed at λmax of the drug in methanol at 229 nm. The calibration curve showed linearity in the range of 1-15 µg/ml concentration with R2 value of 0.998. To evaluate the solubility of meclizine hydrochloride, saturated solutions were prepared in different solvents, PG, PEG 400, PEG 200, PEG 600, glycerine, Tween® 20, Tween® 80, 0.01 N HCl and water. For that, initially 10 mg of drug was added in to 2 g of solvent in Eppendorf tube and closed tubes were shaken for 48 h at 25 °C on a temperature-controlled orbital shaker (Remi Equipments, India). If the drug is dissolved, further drug was added in increments of 10 mg until saturated solution is obtained. After the shaking period, the saturated solutions were centrifuged at 1000 rotations per minute (rpm) for 5 min using a Remi Equipments centrifuge. The resulting supernatant was filtered through a 0.45 µm filter. The 1g of supernatant was diluted with 5 ml of methanol. Further dilutions were made if absorbance reading was out of the range of calibration curve. The solubility of the drug was analyzed using a UV/Visible Spectrophotometer [17, 18]. Each experiment was performed in triplicates.
Drug-excipient compatibility study
Before developing formulations, drug-excipient compatibility study was conducted. For that drug and individual excipients were mixed in the ratio of 1:1 (100 mg: 100 mg) in a glass vial. Closed glass vials were kept in a stability chamber (LSCG–100, Electrolab) at 40 ℃ and 75% relative humidity for 4 w. Closed glass vial with pure drug was also kept as a control sample. Possible interactions between drug and excipients by checked by Fourier Transform Infrared (FTIR) using Bruker® Alpha II FTIR spectrophotometer [19].
Formulation of liquisolid compacts
For liquisolid compact preparation, total 50 tablets batch was calculated. The selected non-volatile water-miscible solvent was used to solubilize the drug. The calculated amount of carrier material, coating material and liquid mixture of drug solution was taken in mortar and mixed thoroughly by pestle manually [20]. The disintegrant sodium starch glycolate and diluent lactose monohydrate were then added to the mortar and mixed for 15 min. The mixture was transferred into polyethylene bag and was mixed for 5 min by tumbling movement done manually. The lubricant magnesium stearate was added at the end and the mixture was mixed for 2 min. The resulting mixture was compressed into tablets of weight 540 mg using 12 mm concave punch at turret speed of 8 rpm using D tooling tablet compression machine (Hardik Engineering, Ahmedabad, India).
Mathematical model for formulation of the liquisolid compact
PG was chosen as the liquid base in this study because the drug shows highest solubility in it. Avicel® PH 102 was used as a carrier material, and Aerosil® 200 was used as a coating material. A mathematical formula helped decide how much of these excipients were needed to make the liquisolid compact [8, 21].
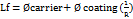 ……. (1)
……. (1)
Here, Lf (liquid load factor) represents the amount of liquid medication added to the carrier and coating materials, Øcarrier represents the liquid retention potential for the carrier material (Avicel® PH 102), Øcoating represents the liquid retention potential for the coating material (Aerosil® 200) and R represents the ratio of the weight of the carrier material to the coating material.
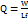 …. (2)
…. (2)
Here, W represents the weight of the liquid medication added, and Q represents the amount of carrier material used in the formulation
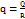 ……. (3)
……. (3)
Here, q represents the weight of coating material used in the formulation
Optimization
The study used a 3² factorial design to understand how two factors-carrier-coating ratio (X1) and drug concentration in the liquid medication (X2)-affected two outcomes: the percentage of drug released in 20 min (Y1) and the angle of repose (Y2). The specific levels of these factors were selected based on earlier trial experiments and are shown in table 1.
Table 1: Selection of dependent and independent variables
| Independent variables | Variable level | ||
| Low(-1) | Medium(0) | High(+1) | |
| Carrier coating ratio (R) (X1) | 23 | 25.5 | 28 |
| Drug concentration in liquid medication (%w/w) (X2) | 10 | 12.5 | 15 |
Table 2 provides the composition of the formulated factorial batches [22]. The formulations of factorial batches were prepared by the method mentioned in the Formulation of liquisolid compacts section.
Table 2: Formulation of factorial batches
| Batch No. | R | Cd (% w/w) | Lf | W (mg) | Q (mg) | Q (mg) | Disintegrant (5%w/w) (mg) | Total weight (mg) |
| F1 | 23 | 10 | 0.37 | 125 | 332.7 | 14.4 | 23.61 | 495.83 |
| F2 | 25.5 | 10 | 0.36 | 125 | 344.9 | 13.5 | 24.17 | 507.67 |
| F3 | 28 | 10 | 0.35 | 125 | 355.6 | 12.7 | 24.66 | 518.06 |
| F4 | 23 | 12.5 | 0.37 | 100 | 266.2 | 11.5 | 18.88 | 396.66 |
| F5 | 25.5 | 12.5 | 0.36 | 100 | 275.9 | 10.8 | 19.33 | 406.13 |
| F6 | 28 | 12.5 | 0.35 | 100 | 284.5 | 10.1 | 19.73 | 414.45 |
| F7 | 23 | 15 | 0.37 | 83.33 | 221.8 | 9.6 | 15.74 | 330.55 |
| F8 | 25.5 | 15 | 0.36 | 83.33 | 229.9 | 9.10 | 16.11 | 338.44 |
| F9 | 28 | 15 | 0.35 | 83.33 | 237.1 | 8.4 | 16.44 | 345.57 |
*R is the ratio of the weight of the carrier material to the coating material, Cd is drug concentration in the liquid medication, Lf is (liquid load factor) represents the amount of liquid medication added to the carrier and coating materials, W is the weight of the liquid medication added, Q represents the amount of carrier material used in the formulation and q represents the weight of coating material used in the formulation.
Characterization part
Pre-compression parameters
The study assessed the pre-compression properties of the liquisolid compacts: bulk density, tapped density, Carr's index, Hausner's ratio, and angle of repose [23]. For bulk density, 30 g of the sample was transferred to a 50 ml cylinder, and its volume was noted. Tapped density was measured using a tapping apparatus until the sample's height stabilized, and the final volume was recorded. These measurements were used to calculate Carr's index and Hausner's ratio. To determine the angle of repose, 20 g of the sample was allowed to flow through a funnel set 2 cm above a flat surface, and the pile's radius was measured to calculate the angle [24]. Each test was conducted in triplicate, and standard deviations were recorded.
Post compression parameters
After the preparation of liquisolid compacts, various post-compression evaluation tests were performed to determine their quality attributes. These tests included the uniformity of weight test, friability test, and disintegration test, as per the guidelines provided by the Indian Pharmacopoeia (IP) 2022. The drug content assay was performed at 229 nm using a UV-visible spectrophotometer with methanol as blank solution. The compacts' hardness was measured using a digital hardness tester. (EiE Instruments Pvt. Ltd., Ahmedabad, India). All the experiments were performed in triplicates and standard deviation was calculated [25].
In vitro drug release
The prepared liquisolid compacts were evaluated for an in vitro dissolution study to analyze their drug release profile. Following the meclizine hydrochloride Tablets IP 2022 guidelines, the study was conducted in 900 ml of 0.01N HCl at 37 °C±0.5 °C, using an IP type II basket apparatus rotating at 100 rpm. Samples were collected at specific time intervals, and the drug release was measured at 231 nm using a UV/Visible Spectrophotometer. The cumulative drug release percentage was calculated and plotted against time to generate the dissolution profile [26, 27]. All experiments were performed in triplicate, and standard deviations were recorded.
DSC analysis
The thermal behaviour of pure meclizine hydrochloride and the optimized formulation was analyzed using DSC equipment (Shimadzu DSC 60, Japan). For this analysis, 50 mg of each sample was accurately weighed, sealed in aluminium pans, and heated from 50 °C to 250 °C at a rate of 10 °C per minute. An empty sealed aluminium pan served as a reference to account for background effects. The resulting DSC spectra were used to examine the thermal properties of the substances [28].
XRD study
Meclizine hydrochloride pure drug and optimized formulation were tested for XRD using an X-ray diffractometer (Xpert Pro MP, Panalytical, The Netherlands) equipped with cobalt radiation with a wavelength of 1.540 Å, a voltage of 40 KV, and a current of 30 mA. The analysis was performed in the 2θ range of 5° to 40° with a scanning rate of 0.1°/min [29].
Stability study
The accelerated stability studies were performed to evaluate the stability of the optimized formulation. To conduct the accelerated stability studies, the optimized formulation was packaged in alu-alu strip package and stored in a stability chamber at a temperature of 40±2 °C and relative humidity (RH) of 75±5% for a period of six months. Sampling of the formulation was conducted at predetermined intervals of 0, 30, 90 and 180 days to assess its stability [30]. After the period of six months, the formulation was evaluated for change in visual appearance, drug assay and dissolution studies.
RESULTS AND DISCUSSION
Solubility study in various solvents
Based on the findings presented in table 3, the saturation solubility of meclizine hydrochloride was found to decrease in the following order: Propylene glycol>PEG 600>Tween® 80>PEG 200>Glycerine>Tween® 20>PEG 400>0.01 N HCl>Water. The researchers selected propylene glycol as the non-volatile solvent in the preparation of the liquisolid compact due to its significantly high solubility [31]. Various other reports of liquisolid compact also used propylene glycol as a non-volatile solvent. Because of higher solubility of drug in propylene glycol, highest drug loading can be achieved in the minimum volume of solvent that will require minimum quantity of carrier and coating material to incorporate non-volatile solvent, which leads to small tablets [32]. Propylene glycol is also in the USFDA generally recognized as safe (GRAS) list; therefore, it is safe for human consumption [33].
Table 3: Solubility study in various solvents
| Solvent | Solubility (mg/g)* |
| Water | 0.0069±0.0006 |
| Propylene glycol | 267.12±0.34 |
| PEG 200 | 217.66 ±0.2 |
| PEG 400 | 155.14±0.3 |
| PEG 600 | 23.63±0.1 |
| Tween® 20 | 55.75±0.74 |
| Tween® 80 | 215.12±0.98 |
| Glycerine | 76.23±0.21 |
| 0.01 N HCL | 0.0082±0.0005 |
*All values are±SD which are mean of three determination
Drug-excipient compatibility study
The FTIR spectra of pure drug and the drug-excipient mixtures were compared. All the characteristic peaks of the drug were present after 4 w of study, which concludes that drug and excipients were compatible [19].
Evaluation of factorial batches
Pre-compression parameters
According to the results presented in table 4, the angle of repose for all the prepared formulations fell within the range of 27.33° to 34.66°. This indicated that batches F1, F4, and F7 had good flow properties, while the other batches had acceptable flow properties. The Carr's index values ranged from 9.3 to 19.56, indicating good to passable flow properties of the formulations. The Hausner's ratio values were all below 1.29, which is considered acceptable for good flow properties.
Table 4: Pre-compression parameters of factorial batches
| Batch code | Bulk density (gm/cm3) | Tapped density (gm/cm3) | Carr’s Index (%) | Hausner’s ratio | Angle of repose (°) |
| F1 | 0.39±0.02 | 0.43±0.01 | 9.3±0.12 | 1.1±0.01 | 27.66±0.57 |
| F2 | 0.36±0.03 | 0.42±0.03 | 14.28±0.16 | 1.16±0.01 | 31.12±0.66 |
| F3 | 0.37±0.01 | 0.46±0.04 | 19.56±0.19 | 1.24±0.02 | 34.66±1 |
| F4 | 0.35±0.04 | 0.4±0.02 | 12.5±0.23 | 1.14±0.01 | 29.16±0.57 |
| F5 | 0.4±0.03 | 0.46±0.01 | 13.04±0.21 | 1.15±0.02 | 31.33±0.57 |
| F6 | 0.36±0.06 | 0.44±0.05 | 18.18±0.14 | 1.22±0.01 | 33.66±1 |
| F7 | 0.36±0.01 | 0.4±0.04 | 10±0.16 | 1.11±0.02 | 27.33±0.66 |
| F8 | 0.35±0.05 | 0.41±0.05 | 14.63±0.28 | 1.17±0.01 | 30.66±0.57 |
| F9 | 0.38±0.04 | 0.45±0.03 | 15.55±0.21 | 1.18±0.02 | 31.27±1 |
*All values are±SD which are mean of three determination
Post compression parameters
Table 5 presents the post-compression parameters of the factorial batches. Uniformity of weight test passed for all the batches. Friability of all the batches was less than 1% limit. Hardness of the batches were kept around 5 kg/cm2. Assay was well within the meclizine hydrochloride tablet assay limit of 95%-105% as per monograph. Batches showed disintegration ranging from 96 sec to 132 sec [34, 35]. Here, the disintegration result for the compact varies from 96.84 to 131.73 sec. It is observed in general that for carrier coating ration (R) value of 23, the disintegration time is more compared to the R-value of 28. Avicel® PH 102 has excellent disintegration property [36] and in formulas with R value of 28, the proportion of Avicel® PH 102 in the overall formula is high. This leads to faster disintegration with R value 28. The friability of the tablets are within the limit but still relatively high. This may be due to liquid entrapment within the formulations. However, suitable strip packaging can be used to minimize the friability as a precaution [37].
Table 5: Post-compression parameters of optimize formulation
| Evaluation parameter | Friability test (%) | Hardness (kg/cm2) | Assay (%) | Disintegration test (sec) |
| F1 | 0.43±0.05 | 5.33±0.70 | 99.81±0.65 | 127.54±3.68 |
| F2 | 0.58±0.10 | 5.66±0.33 | 98.98±0.66 | 123.74±4.43 |
| F3 | 0.51±0.02 | 5.23±0.83 | 99.43±0.45 | 98.21±1.66 |
| F4 | 0.62±0.03 | 5.52±0.64 | 98.43±0.82 | 106.44±5.53 |
| F5 | 0.38±0.01 | 5.18±0.94 | 100.54±0.69 | 119.63±3.69 |
| F6 | 0.26±0.00 | 6.16±0.84 | 99.46±0.57 | 96.84±3.72 |
| F7 | 0.57±0.03 | 5.29±1.26 | 99.53±0.48 | 131.73±4.82 |
| F8 | 0.46±0.03 | 5.85±0.93 | 98.52±1.64 | 116.84±6.82 |
| F9 | 0.38±0.02 | 5.59±0.27 | 99.64±0.84 | 104.58±7.55 |
*All values are±SD which are mean of three determination
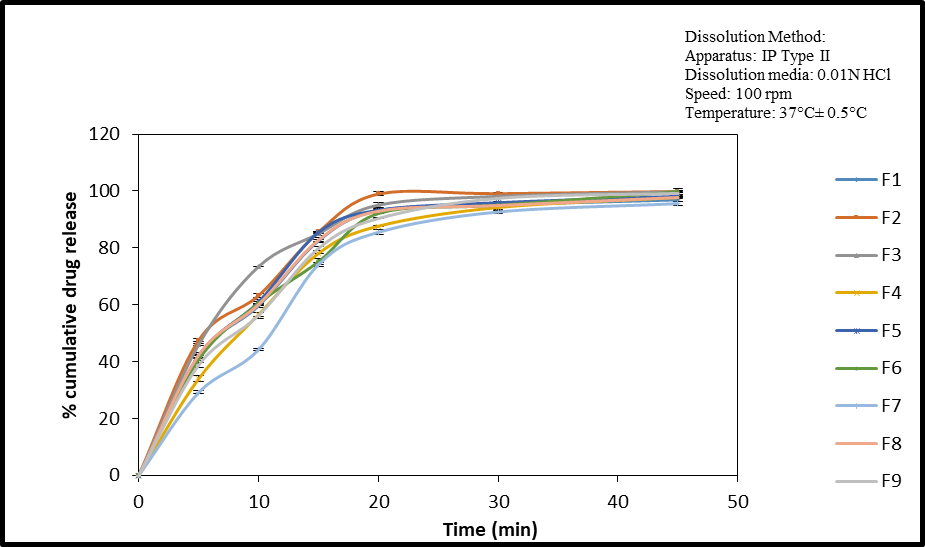
Fig. 1: % cumulative drug release versus time where batches F2 and F3 show faster dissolution compared to batches F7 and F9 (n=3)
In vitro dissolution profile for factorial batches
The drug release results obtained at different time intervals were plotted against time to generate the dissolution profiles, as shown in fig. 1.
Optimization process
Statistical analysis of 32 factorial design
It was observed that the best-fit model was quadratic model and quadratic polynomial equation for responses Y1, Y2 is given here: Y1=93.96+1.87X1-3.055X2+0.72X1X2-4.41X12+1.63X22 and Y2=31.50+2.34X1-0.79X2-0.61X1X2-0.18X12-0.7X22. ANOVA for the above models were calculated using a confidence interval 95% and p-values were found to be 0.00422 and 0.0140 respectively for Y1, Y2. p values are less than 0.05 which concludes that the models are statistically significant. The observed value for cumulative % drug release at 20 min (Y1) for batches F1-F9 varied from 85.51-98.95 %. Summary of multiple regression analysis for Y1, Y2 has been given in table 6. It is observed that Y1 (Cumulative % drug release at 20 min) is positively affected by X1 and negatively by X2. However, the interaction effect of X1X2 is not significant. For Y2 (Angle of repose), X1 has a positive effect and X2 has a negative effect. For Y2 also, there is no significant interaction effect.
Table 6: Summary of results of multiple regression analysis for Y1, Y2
| Dependent variables | Y1(Cumulative % drug release at 20 min) | Y2(Angle of repose) | ||
| Coefficients | P value | Coefficients | P value | |
| Intercept | 93.96 | 0.0042 | 31.50 | 0.0141 |
| X1 | 1.87 | 0.0071 | 2.34 | 0.0024 |
| X2 | -3.055 | 0.0017 | -0.79 | 0.0461 |
| X1X2 | 0.72 | 0.130 | -0.61 | 0.129 |
| X12 | -4.416 | 0.0029 | -0.18 | 0.696 |
| X22 | 1.638 | 0.044 | -0.7 | 0.193 |
Contour plots and response surface analysis
Based on the three-dimensional plot shown in fig. 2, an increase in the carrier-coating ratio (R) resulted in an increase in the % cumulative drug release at 20 min. As the carrier-coating ratio is increased, proportional quantity of coating material is decreased. Therefore, % cumulative drug release is increased. However, an increase in the drug concentration in the liquid medication (%Cd) led to a decrease in cumulative drug release at 20 min, maybe due to drug precipitation in a smaller volume of liquid. It was also observed that a higher carrier-coating ratio (R) led to a decrease in cumulative drug release at 20 min, which could be attributed to the slightly hydrophobic nature of the carrier and coating materials [38].
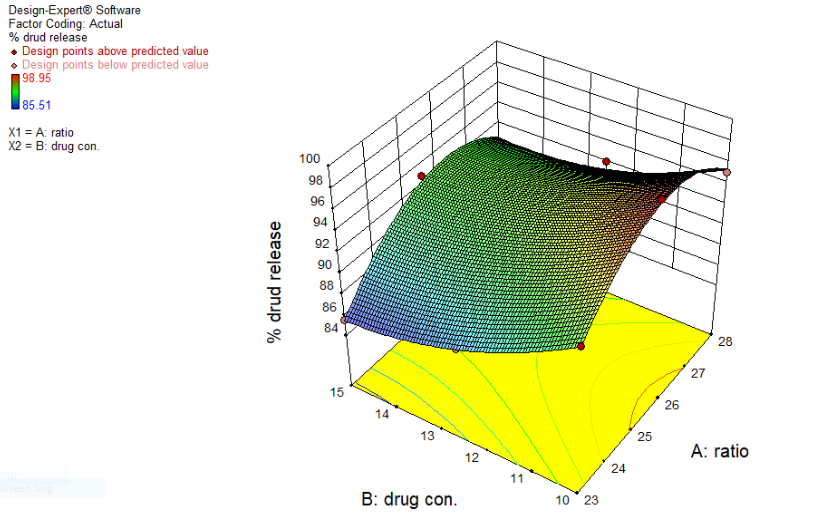
Fig. 2: 3D surface plot showing of X1 and X2 on Y1 where an increase in the carrier-coating ratio (R) resulted in increase in the % cumulative drug release
A 3D plot, as shown in fig. 3, indicates that an increase in the carrier-coating ratio (R) results in an increase in the angle of repose. Conversely, an increase in the drug concentration in the liquid medication causes a decrease in the angle of repose [39].
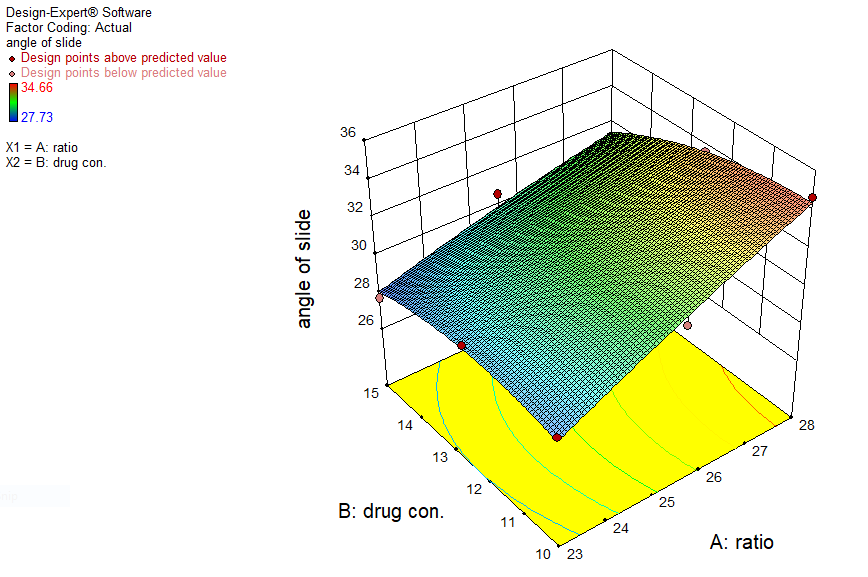
Fig. 3: 3D surface plot showing effect of X1 and X2 on response Y2 where an increase in the carrier-coating ratio (R) results in an increase in the angle of repose
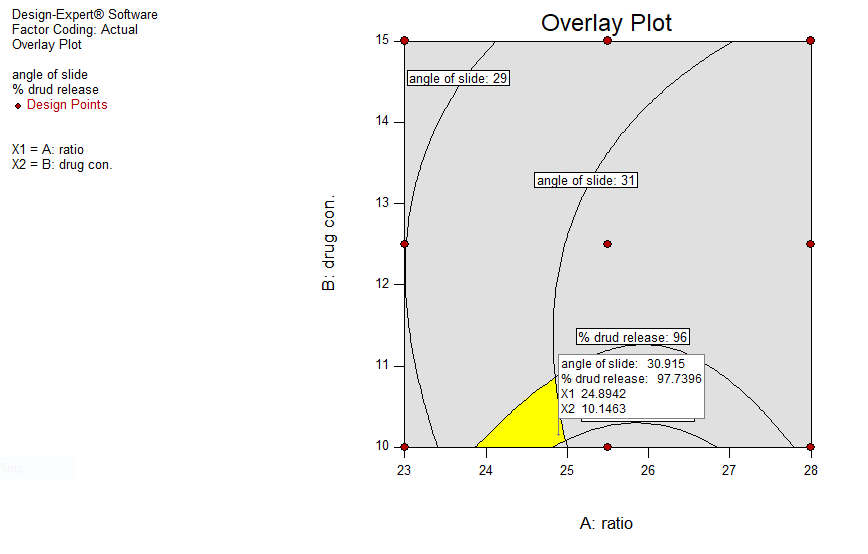
Fig. 4: Overlay plot for O1 where yellow region shows the optimized region
Model validation and selection of optimized batch
Using Design Expert 10.0.1.0 software, the optimized batch (O1) was identified out of optimized region. To validate the design model, formulation O1 was prepared and dependent parameters Y1 and Y2 were evaluated and compared with predicted values, as shown in fig. 4. For Y1 and Y2, the Relative Standard Deviation (RSD) values for predicted vs actual were 1.21% and 1.64% respectively, which is less than 2%. Thus, we can conclude that the statistical model is mathematically valid [40].
Fig. 4 shows the preparation of the optimized batch (O1), with the yellow region representing the optimized area. Table 7 shows the optimized formula for the optimized batch.
DSC study
The thermal properties of pure meclizine hydrochloride and liquisolid compact were analyzed and presented in fig. 5 and 6, respectively. A characteristic endothermic peak at 225.06 °C was observed in the DSC of the pure drug, indicating its crystalline nature. However, in the DSC of liquisolid compact, there was no peak observed at 225.06 °C, suggesting that the drug might have been completely converted into an amorphous form or solubilized in the prepared system, which could have contributed to the enhanced dissolution [26-28].
Table 7: The formula for optimized and checkpoint batches
| Ingredient | Quantity (mg) |
| O1 | |
| Meclizine hydrochloride | 15.41 (Equivalent to 12.5 mg of meclizine) |
| PG | 123 |
| Carrier (Avicel® PH 102) | 341.66 |
| Coating (Aerosil® 200) | 13.72 |
| Lactose monohydrate | 19.60 |
| SSG | 23.91 |
| Magnesium stearate | 2.7 |
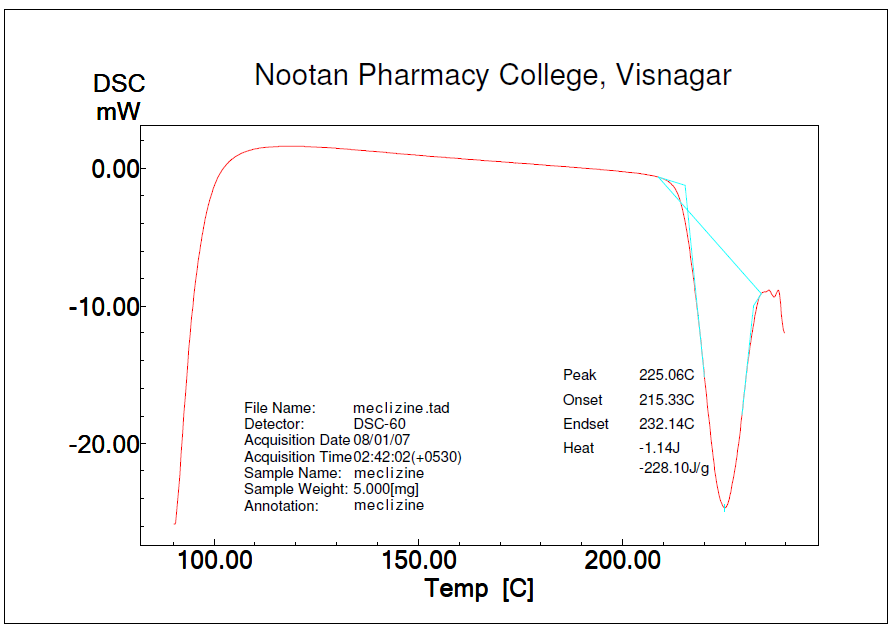
Fig. 5: DSC of meclizine hydrochloride shows sharp endothermic peak of pure drug at 225.06 °C
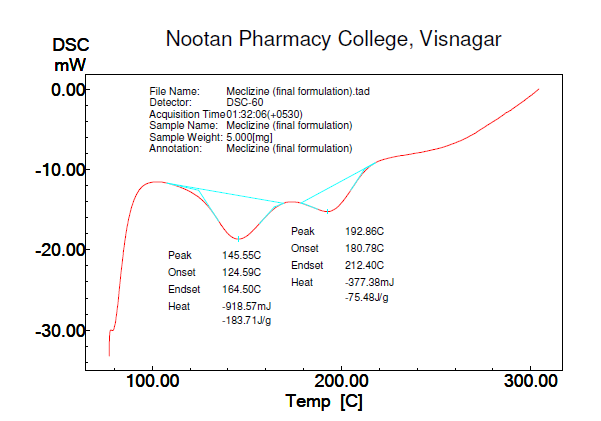
Fig. 6: DSC of final formulation shows an absence of peak at 225.06 °C
XRD analysis
The X-ray diffraction spectra of meclizine hydrochloride drug and the optimized formulation (O1) are presented in fig. 7. The XRD pattern of pure meclizine hydrochloride showed sharp peaks at specific 2θ diffraction angles, indicating its crystalline nature. However, the optimized liquisolid formulation showed an absence of sharp peaks in its XRD pattern, suggesting the lack of crystallinity. This could be attributed to the complete conversion of meclizine hydrochloride from its crystalline to solubilized form resulting from its solubilization in the liquid vehicle used in the liquisolid preparation [28].
Comparison of cumulative drug release from optimized batch with pure drug
The results depicted in fig. 8 reveal that the optimized batch exhibited a significantly higher cumulative drug release of 95.54% in 20 min, whereas the pure drug demonstrated a much lower cumulative drug release of only 30.41% in the same time frame. This faster dissolution of the drug can be attributed to its pre-dissolved state in a non-volatile water-soluble liquid, namely Propylene Glycol. Such a state promotes faster wetting, thereby facilitating rapid dissolution of the drug in the medium [23, 28, 32].
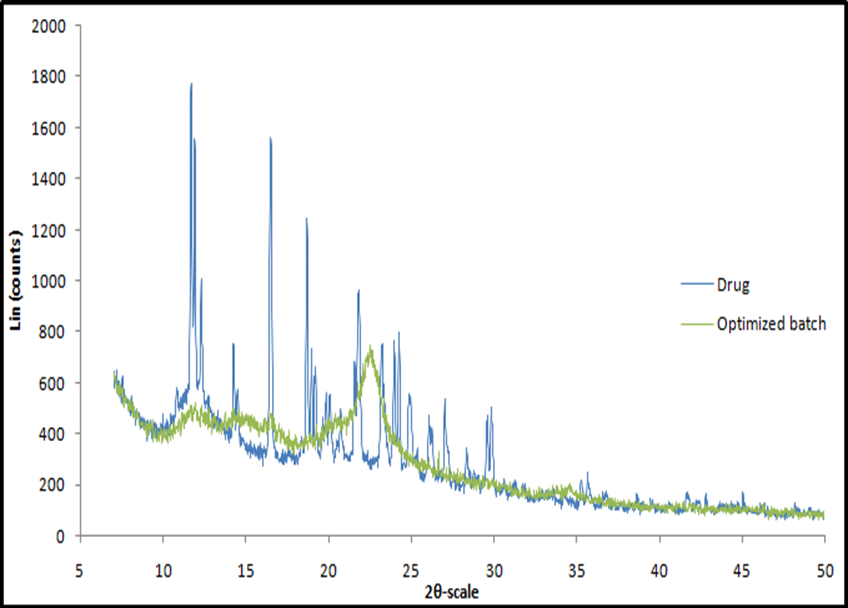
Fig. 7: XRD of meclizine hydrochloride and optimized formulation (O1)
Balance between flowability and dissolution rate in liquisolid compact
The optimum formula of liquisolid compact must establish a balance between flowability and dissolution rate. Flowability is an essential property of powder to achieve uniformity of weight and hopper flow. As per the results, an increase in the drug concentration in the liquid medication (%Cd) led to a decrease in cumulative drug release (fig. 2). That effectively means more amount of water miscible vehicle leads to faster dissolution rate. But, an increase in amount of water-miscible vehicle in the formula results in an increased angle of repose – a measure of decreased flow (fig. 3). This happens because the powder to be compacted can incorporate a limited amount of vehicle where the flow is acceptable, beyond which the flow property of the powder becomes un-acceptable due to higher liquid amount. Therefore, to deliver a dose of drug (which cannot be changed), optimized concentration and, therefore, amount of vehicle must be chosen so that there remains a balance between flowability and dissolution rate [10-12].
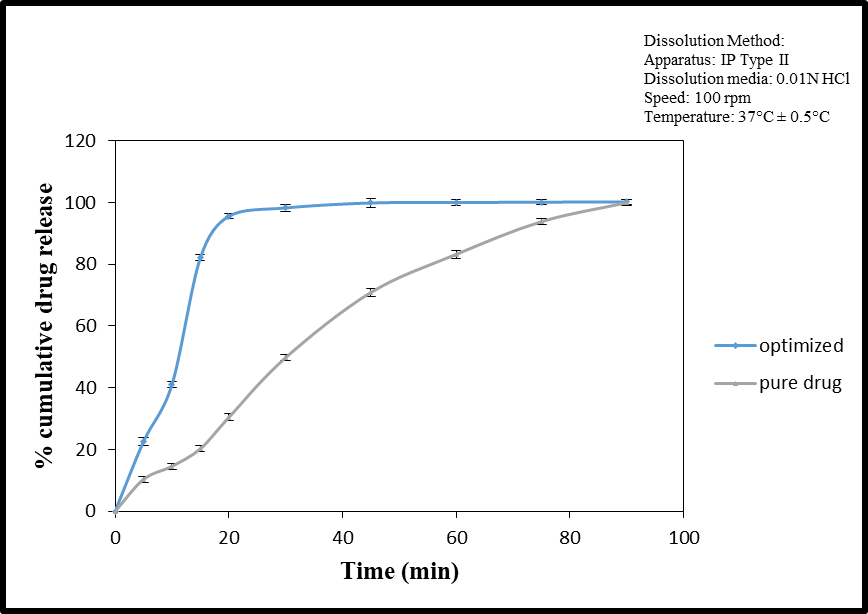
Fig. 8: Comparison of % cumulative drug release of optimized batch with pure drug shows significantly rapid dissolution of the drug in optimized batch (n=3)
Stability study
After conducting a stability study on the optimized batch, it was discovered that there were no noteworthy changes in the quality parameters of the formulation even after six months under accelerated stability conditions, which indicates that the product is stable.
CONCLUSION
For developing meclizine hydrochloride liquisolid compacts, solubility of the drug was assessed in various water-miscible solvents. Among these, propylene glycol was found to be the solvent with the highest solubility. Avicel® PH 102 was selected as the carrier and Aerosil® 200 as the coating material. The optimization of the formulation was carried out using Design Expert 10.0.1.0 software and a 32 factorial design was employed. The results indicated that an increase in the ratio of carrier coating decreased flow property while increasing cumulative % cumulative drug release at 20 min. additionally, an increase in drug concentration in the liquid medication decreased % cumulative drug release at 20 min while increasing flow property. Accelerated stability study exhibited no significant change in physical appearance, % drug content, or % cumulative drug release after six months. These findings suggest that a liquisolid formulation could be a promising alternative for the faster drug release and dissolution enhancement of meclizine hydrochloride. However, a liquisolid tablet of meclizine hydrochloride has more number of excipients, more processing steps and significantly higher weight than a conventional tablet for same dose. Due to multiple additional processes and additional cost of non-volatile solvents, it is challenging to develop an industrially scalable and commercially viable liquisolid tablet of meclizine hydrochloride. Current study has only studied in vitro aspects of the formulation. In future, the formulation can be evaluated in animals for assessing bioavailability and can be extended to bioavailability clinical trials in humans compared to conventional tablets.
FUNDING
Nil
AUTHORS OF CONTRIBUTIONS
Tularam Barot: Conceptualization, Literature review, Experimental part, Data analysis and Writing original draft; Karuna Nagula: Writing the manuscript, critical editing; Moksha Patel: Literature review, Experimental part, Data analysis; l D Patel: Writing the manuscript, critical editing
CONFLICT OF INTERESTS
Declared none
REFERENCESReferences
Darwesh AY, El Dahhan MS, Meshali MM. A new dual-function orodissolvable/dispersible meclizine HCl tablet to challenge patient inconvenience: in vitro evaluation and in vivo assessment in human volunteers. Drug Deliv Transl Res. 2021;11(5):2209-23. doi: 10.1007/s13346-020-00889-z, PMID 33443718.
Qazi F, Shoaib MH, Yousuf RI, Siddiqui F, Nasiri MI, Ahmed K. QBD based eudragit coated meclizine HCl immediate and extended-release multiparticulates: formulation characterization and pharmacokinetic evaluation using HPLC-Fluorescence detection method. Sci Rep. 2020;10(1):14765. doi: 10.1038/s41598-020-71751-y, PMID 32913337.
Vemula SK, Vangala M. Formulation development and characterization of meclizine hydrochloride sublimated fast dissolving tablets. Int Sch Res Not. 2014;2014:281376. doi: 10.1155/2014/281376, PMID 27355021.
Wang Z, Lee B, Pearce D, Qian S, Wang Y, Zhang Q. Meclizine metabolism and pharmacokinetics: formulation on its absorption. J Clin Pharmacol. 2012;52(9):1343-9. doi: 10.1177/0091270011414575, PMID 21903894.
George SJ, Vasudevan DT. Studies on the Preparation characterization and solubility of 2-HP-β-Cyclodextrin meclizine HCl inclusion complexes. J Young Pharm. 2012;4(4):220-7. doi: 10.4103/0975-1483.104365, PMID 23493156.
Meclizine. American society of Health-System Pharmacists; c 2024. Available from: https://medlineplus.gov/druginfo/meds/a682548.html. [Last accessed on 2 Jan 2025].
Jaydip B, Dhaval M, Soniwala MM, Chavda J. Formulation and optimization of liquisolid compact for enhancing dissolution properties of efavirenz by using DoE approach. Saudi Pharm J. 2020;28(6):737-45. doi: 10.1016/j.jsps.2020.04.016, PMID 32550806.
Spireas SS, Jarowski CI, Rohera BD. Powdered solution technology: principles and mechanism. Pharm Res. 1992;9(10):1351-8. doi: 10.1023/a:1015877905988, PMID 1448438.
Elkhodairy KA, Elsaghir HA, Al Subayiel AM. Formulation of indomethacin colon targeted delivery systems using polysaccharides as carriers by applying liquisolid technique. Bio Med Res Int. 2014;2014:704362. doi: 10.1155/2014/704362, PMID 24971345.
Baby JN, Manjila SB, Bijin EN, Constantine I, Pramod K, Valsalakumari J. Design and technology of liquisolid compacts. J App Pharm Sc. 2013;38 Suppl 1:S111-9. doi: 10.7324/JAPS.2013.38.S17.
Gajdziok J, Vranikova B. Enhancing of drug bioavailability using liquisolid system formulation. Ceska Slov Farm. 2015;64(3):55-66. doi: 10.36290/csf.2015.018, PMID 26400228.
LU M, Xing H, Jiang J, Chen X, Yang T, Wang D. Liquisolid technique and its applications in pharmaceutics. Asian J Pharm Sci. 2017;12(2):115-23. doi: 10.1016/j.ajps.2016.09.007, PMID 32104320.
Nagaich U. Pharmaceutical applications of liquisolid technique. J Adv Pharm Technol Res. 2018;9(2):43. doi: 10.4103/japtr.JAPTR_236_18, PMID 30131935.
Bhide P, Nachinolkar R. Formulation development and characterisation of meclizine hydrochloride fast dissolving tablets using solid dispersion technique. Int J App Pharm. 2018;10(4):141-6. doi: 10.22159/ijap.2018v10i4.26493.
Sisinthy SP, Selladurai S. Cinnarizine liquid solid compacts: preparation evaluation. Int J App Pharm. 2019;11(1):150-7. doi: 10.22159/ijap.2019v11i1.30109.
Pathak BK, Raghav M, Thakkar AR, Vyas BA, Shah PJ. Enhanced oral bioavailability of etodolac by the liquisolid compact technique: optimisation in vitro and in vivo evaluation. Curr Drug Deliv. 2021;18(4):471-86. doi: 10.2174/1567201817666201026111559, PMID 33106143.
Chella N, Shastri N, Tadikonda RR. Use of the liquisolid compact technique for improvement of the dissolution rate of valsartan. Acta Pharmaceutica Sinica B. 2012;2(5):502-8. doi: 10.1016/j.apsb.2012.07.005.
Rede SD, Jani RK. Solubility enhancement of paclitaxel by using biomaterial solubility enhancement of paclitaxel by using biomaterial. Res J Pharm Technol. 2022;15(11):5089-3.
Margret S, Blr M. Study of direct compression method for the preparation of quinapril hydrochloride tablets. Asian J Pharm Clin Res. 2020;13(1):202-11.
Spireas S, Bolton SM. Liquisolid systems and methods of preparing same. United States Patent US5800834A; 1996.
Hentzschel CM, Alnaief M, Smirnova I, Sakmann A, Leopold CS. Enhancement of griseofulvin release from liquisolid compacts. Eur J Pharm Biopharm. 2012;80(1):130-5. doi: 10.1016/j.ejpb.2011.08.001, PMID 21846502.
Panda S, Varaprasad R, Priyanka K, Swain RP. Liquisolid technique: a novel approach for dosage form design. Int J App Pharm. 2017;9(3):8-14. doi: 10.22159/ijap.2017v9i3.18698.
Patel DS, Pipaliya RM, Surti N. Liquisolid tablets for dissolution enhancement of a hypolipidemic drug. Indian J Pharm Sci. 2015;77(3):290-8. doi: 10.4103/0250-474x.159618, PMID 26180274.
Patric JS. Martins physical pharmacy and pharmaceutical sciences. 2nd ed. New Delhi: Wolters Kluwer (India) Pvt. Ltd; 2008.
Naureen F, Shah Y, Shah SI, Abbas M, Rehman IU, Muhammad S. Formulation development of mirtazapine liquisolid compacts: optimization using central composite design. Molecules. 2022;27(13):4005. doi: 10.3390/molecules27134005, PMID 35807252.
Gao Z, YU L, Clark S, Trehy M, Moore T, Westenberger B. Dissolution testing for bioavailability of over-the-counter (OTC) drugs a technical note. AAPS Pharm Sci Tech. 2015;16(5):1227-33. doi: 10.1208/s12249-015-0297-x, PMID 25680355.
Dalvadi H, Patel J, Shah D. Time and/or site-specific drug delivery of floating pulsatile release delivery system. Syst Rev Pharm. 2011;2(1):59-65. doi: 10.4103/0975-8453.83441.
Molaei MA, Osouli Bostanabad K, Adibkia K, Shokri J, Asnaashari S, Javadzadeh Y. Enhancement of ketoconazole dissolution rate by the liquisolid technique. Acta Pharm. 2018;68(3):325-36. doi: 10.2478/acph-2018-0025, PMID 31259692.
Jaydip B, Dhaval M, Soniwala MM, Chavda J. Formulation and optimization of liquisolid compact for enhancing dissolution properties of efavirenz by using DoE approach. Saudi Pharm J. 2020;28(6):737-45. doi: 10.1016/j.jsps.2020.04.016, PMID 32550806.
Cirri M, Mura P, Valleri M, Brunetti L. Development and characterization of liquisolid tablets based on mesoporous clays or silicas for improving glyburide dissolution. Pharmaceutics. 2020;12(6):503. doi: 10.3390/pharmaceutics12060503, PMID 32492869.
Dalal L, Allaf AW, El Zein H. Formulation and in vitro evaluation of self-nanoemulsifying liquisolid tablets of furosemide. Sci Rep. 2021;11(1):1315. doi: 10.1038/s41598-020-79940-5, PMID 33446749.
Sura RS, Subrahmanyam C, Rachamalla SS. Design and evaluation of liquisolid compacts of nebivolol hydrochloride. Int J App Pharm. 2022;14(2):293-307. doi: 10.22159/ijap.2022v14i2.43657.
Generally recognized as safe (GRAS). United States Food and Drug Administration; c2024. Available from: https://www.fda.gov/food/food-ingredients-packaging/generallyrecognizedsafe-gras. [Last accessed on 03 Jan 2025].
Ministry of health and family welfare. Indian pharmacopoeia. 1st ed. Ghaziabad: The Indian Pharmacopoeia Commission; 2022.
Vaskula S, Vemula SK, Bontha VK, Garrepally P. Liquisolid compacts: an approach to enhance the dissolution rate of nimesulide. J Appl Pharm Sci. 2012;2(5):115-21.
Nair HA, Gadhiraju G, Sunny G. Development of orodispersible tablets of loratadine containing an amorphous solid dispersion of the drug in soluplus® using design of experiments. Int J Pharm Pharm Sci. 2023;15(8):19-27. doi: 10.22159/ijpps.2023v15i8.47750.
Pai GK, Krishna V, Reddy S, Kishan YS, Aditya V, Bhanuprakash G. Physical evaluation of the uncoated tablets of glimepiride with different types of primary packaging. Res J Pharm Technol. 2012;5(3):404-7.
Oza N, Sahoo S, Sagar S. A 32 full factorial design for topical controlled releasetazarotene microsponge using HPMC gel. Int J App Pharm. 2019;11(5):12-8. doi: 10.22159/ijap.2019v11i5.34496.
Saeedi M, Akbari J, Morteza Semnani K, Enayati Fard R, Sar Reshteh Dar S, Soleymani A. Enhancement of dissolution rate of indomethacin: using liquisolid compacts. Iran J Pharm Res. 2011;10(1):25-34. PMID 24363677.
Bose A, Wong TW, Singh N. Formulation development and optimization of sustained release matrix tablet of itopride HCl by response surface methodology and its evaluation of release kinetics. Saudi Pharm J. 2013;21(2):201-13. doi: 10.1016/j.jsps.2012.03.006, PMID 23960836.