
Int J App Pharm, Vol 17, Issue 3, 2025, 148-156Review Article
REVOLUTIONIZING DRUG DEVELOPMENT: HOW AI AND MACHINE LEARNING ARE SHAPING THE FUTURE OF MEDICINE-A REVIEW
GIRIDHARA M.1, GURUBARAN S.1, SANJAI RAJAGOPAL1, GOWTHAM ANGAMUTHU2, NAGASAMY VENKATESH DHANDAPANI1*
1Department of Pharmaceutics, JSS College of Pharmacy, Ooty, Nilgiris, Tamil Nadu, India. 2Department of Pharmaceutical Regulatory Affairs, JSS College of Pharmacy, Ooty, Nilgiris, JSS Academy of Higher Education and Research, Mysuru, Tamil Nadu, India
*Corresponding author: Nagasamy Venkatesh Dhandapani; *Email: nagasamyvenkatesh@jssuni.edu.in
Received: 18 Dec 2024, Revised and Accepted: 21 Feb 2025
ABSTRACT
AI and Machine Learning are revolutionizing drug development, which was previously characterized as being complicated, time-consuming, and costly. Through the application of enormous biomedical data and high-performance computing capabilities, AI/ml algorithms speed up different aspects of drug discovery, such as target identification, lead optimization, and clinical trial design. The technologies uncover intricate biological patterns, making it possible to have personalized treatments and more effective drug targeting at the bedside.
AI methods have demonstrated phenomenal success in protein structure prediction, virtual compound screening, and de novo drug design, shortening project duration from years to weeks. Some of the highlights are Alpha Fold's record-breaking accuracy in protein structure predictions by DeepMind and Deep Tox's 86% accuracy for predicting toxicity. Neural networks have reached 92% accuracy in predicting protein-protein interactions, while AI integrated with organ-on-chip systems has cut down early-stage drug screening time by 60% and enhanced prediction accuracy by 40%. Joint efforts such as melloddy allow the pharmaceutical industry to collaborate without compromising data privacy and security measures.
However considerable issues remain around data quality, interpretability of models, and regulation compliance in the pharma industry. Advances in AI in the future, fueled by cross-disciplinary cooperation and ongoing technological progress, will play a vital role in surmounting these issues and making AI/ml bring about revolutionary changes in drug discovery to make therapies more available and affordable to global patients. The science keeps on unfolding, tipping the balance between potential transformation and built-in complexities in pharmaceutical usage.
Keywords: Artificial intelligence, Machine learning, Drug development, Preclinical studies, Clinical studies
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i3.53457 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
The pharmaceutical industry stands at a crossroads. New healthcare technologies are piling up new challenges on the horizon in the development of drugs. Indeed, the development process has always been notoriously complex, time-consuming, and cost-intensive. Recent estimates place the average cost of developing a new drug at about $2.6 billion-a period that often surpasses over a decade from discovery in the laboratory to approval for drug marketing [1]. High attrition rates exacerbate the huge time and resources poured into such a pipeline of which an almost infinitesimally small proportion of candidate drugs successfully traverse the very long path from preclinical experiments to getting approvals.
In this space, the hopes of Artificial intelligence and Machine learning in the drug development process stand since these challenges occur. These quickly emerging technologies open big potential to transform several elements of the drug discovery and development pipeline from target identification and lead optimization to clinical trial design and patient stratification [2]. The use of very large biomedical data quantities and more effective algorithms enable Artificial Intelligence and Machine Learning methods to potentially unveil patterns and insights overlooked or untouched by human researchers, hence accelerating the drug development process and also the chance of success.
Research and development for pharmaceuticals can greatly benefit from various sophisticated language models, through the analysis of intricate datasets, prediction of interactions, and identification of possible lead compounds, these AI models speed up the drug discovery and development process. They optimize clinical trials and medication safety monitoring, improve regulatory compliance, and increase the effectiveness of literature reviews. AI-driven platforms have developed into useful instruments for innovation and the creation of new delivery methods in medication delivery research [3].
The application of Artificial Intelligence in drug discovery is not entirely new, with early efforts dating back to the 1960s when pioneering systems like DENDRAL first attempted to automate molecular structure elucidation [4]. The new wave of drug development majorly relies on the application of Artificial Intelligence/Machine Learning in conjunction with recent advances in computational power and explosive growth in biomedical data. Integration of AI with multi-omics data has transformed our understanding of disease mechanisms and drug targets. Modern sequencing technologies produce more than 15 petabytes of genomic data annually, which AI systems can analyze to identify novel drug targets with accuracy rates of 85-90%. This is a huge improvement over the traditional approaches that could only reach an accuracy rate of 30-40% in target identification. Advanced machine learning approaches, especially deep learning, have yielded incredible success in attacking challenging biological problems. One such challenge for the structural biology community to pursue with enormous prospects of drug design has been very successful by DeepMind's Alpha Fold in predicting the protein structure [5].
The influence of Artificial Intelligence and Machine Learning on drug development can go beyond mere efficiency improvements: these technologies may also change the perspective on the understanding of diseases and therapeutic interventions. In principle, it should be possible for Artificial intelligence systems to generate new hypotheses based on the complex analysis of the biological network and types of data disregarded so far as putative drug targets from genomics and proteomics through clinical records and literature [6]. This approach is therefore a particularly promising development for challenging therapeutic areas such as rare diseases or disorders with complex genetic underpinnings.
More importantly, though, Artificial Intelligence and Machine Learning applications in drug development fit along the general trend of precision medicine. These technologies could eventually open the way for better and more targeted therapy because they can eventually help in better stratification, analyzing large-scale patient data, such as genomic, proteomic, and clinical information, enabling AI systems to identify patient subgroups with maximum accuracy, thereby making targeted therapeutic approaches more likely and allow the development of more personalized treatment strategies [7]. This approach can change the outcome and enhance results by patients, optimize the clinical trial success rates by 30-40%, and reduce the overall cost by up to 25%, and time to develop drugs [8]. In addition, effective exploitation of Artificial Intelligence/Machine Learning approaches requires contribution from an infusion of knowledge from a variety of disciplines: biology, chemistry, computer science, and clinical medicine.
The applied reviews critiqued applications and methodologies along with the implications of AI and ML in drug development. Here, a comprehensive overview of the latest status of these developments is presented. Crucial areas where these technologies have a lot to say are addressed, case study analyses of successful implementations are viewed and hints about challenges and futures in these fields are provided. We comment here on the possibility and the limits of Artificial Intelligence/Machine learning in drug development and hence contribute to the continuous discussion on how these powerful tools may best be exploited against urgent healthcare needs that will speed the development of lifesaving therapies.
The selections of articles for the present review were searched from specialized databases (Range of years: 2014-2024) such as Elsevier, Pubmed, and Cambridge using the keywords Artificial Intelligence, Machine Learning, Drug Development. Other selections include articles from Springer Wiley, information from Internet sources, and online published articles from The Lancet Respiratory Medicine, Medscape, and Statpearls.
AI/ml applications in drug discovery
For most types of drug discovery activities, artificial intelligence and machine learning can impact everything from the identification of a target to the lead optimization of a drug and all things in between. The following section embodies this in some of the areas influenced by Artificial Intelligence and Machine Learning.
Target identification and validation
Some of the most fundamental steps of drug discovery would relate to identifying and validating drug targets. Artificial Intelligence and Machine Learning are emerging as tremendous tools in this space that can analyze large, complex biological datasets for potential therapeutic targets.
Most appealing is the very analysis of multi-omics data. Coming shortly will be evidence of the great efficacy of machine learning algorithms and deep models in integrating different types of data, such as genomics, proteomics, and metabolomics. For example, a graph neural network model by Zitnik et al. was created to predict protein-protein and drug-target interactions through multiple biological networks [9]. This approach not only identified novel drug targets but also provided insights into the underlying biological mechanisms of diseases.
The integration of data from multi-omics using the AI/ml approach has helped to revolutionize target discovery to be able to analyze genomics, transcriptomics, proteomics, and even metabolomics information in a multidimensional way. The significant work of Zitnik et al. elucidated how, with graph neural networks, highly complex interactions could be modeled by different biological entities. Their model processes multiple layers of biological networks simultaneously, protein-protein interaction, gene co-expression networks, and drug-target interactions. The multi-modal approach demonstrated an impressive 15% better prediction accuracy compared to the traditional single-omics approaches.
The success of AlphaFold marks a paradigm shift in structural biology and target identification. The architecture involves a deep learning design that can operate on multiple sequence alignments and evolutionary information to predict protein structures at previously unattainable accuracy. Its key mechanism relies on attention-based neural networks focused on the recognition of critical residue-residue contacts and on evolutionary covariance patterns. This breakthrough has accelerated target validation by providing accurate structural information for previously uncharacterized proteins, reducing the time required from months or years to days.
Artificial Intelligence-based methods made excellent promises in the area of target validation and prediction of probabilities for proteins with their potential relevance to drug applications. In one exemplary work, Jastrzębski et al. trained a random forest model to predict the druggability of a protein based on many physicochemical and structural properties [10]. Their model outperformed traditional approaches, offering a more rapid and cost-effective method for prioritizing potential drug targets.
The development of AlphaFold, conceived at DeepMind, represents a really major point of landmark application of Artificial intelligence in structural biology for drug target identification. The new capability to predict protein structures from amino acid sequences allows new areas in understanding the function of proteins and potential sites for molecule binding involved in drug action to be opened [5]. This breakthrough has profound implications for target identification and validation, potentially accelerating the early stages of drug discovery.
Virtual screening and lead optimization
Artificial Intelligence and Machine Learning technology have made tremendous advancements in the technology of virtual screening, which can be conceived as the computational method of identifying promising compounds from huge chemical libraries. Advanced algorithms can browse millions of compounds and enable the prediction of properties and potential efficacy increasingly accurately.
AI-driven methods have replaced conventional virtual screening, marking a paradigm change in accuracy and efficiency. AI-powered platforms can screen billions of compounds in similar amounts of time, whereas traditional virtual screening approaches generally process 106 compounds every day. The procedures and results of the two systems differ significantly when compared which is shown in the fig. 1.
Modern AI-driven virtual screening has achieved tremendous efficiency gain over the conventional approaches. Where conventional high-throughput screening often deals with only 10⁴-10⁵ compounds per week, deep learning models can screen millions of compounds within hours. Stokes et al.'s deep neural network screened more than 107 million molecules from the ZINC15 database to identify novel antibiotic candidates that conventional approaches missed. The model could reduce false positives by 90% compared to the traditional virtual screening methods.
The sequential processes of structure-based filtering, molecular docking, and scoring are crucial to traditional virtual screening; although meticulous, these stages may be computationally and time-intensive. AI-driven methods, on the other hand, use deep learning models that have been trained on large databases of known drug-target interactions. This allows for the quick parallel screening of compounds against several parameters at once.
Perhaps one of the most outstanding examples is that recently reported from Discoidin Domain Receptor1 kinase inhibitors by Insilco Medicine. Their AI system, combining Generative Adversarial Networks with reinforcement learning, was able to design novel inhibitors in merely 21 days. Most promising was one of the lead compounds, with high potency and selectivity, as validated experimentally. This indeed represents a drastic acceleration when compared with the time traditionally observed to reach comparable achievements through standard design approaches, which usually take 2-3 y [11]. Their model, trained on a diverse set of molecules, successfully identified a compound that showed broad-spectrum antibiotic activity against a wide range of pathogens-even some antibiotic-resistant strains. That exciting work shows the power of artificial intelligence, which not only accelerates the screening process but, even more so, the possibility of revealing novel chemical entities-things that otherwise would slip through with traditional approaches.
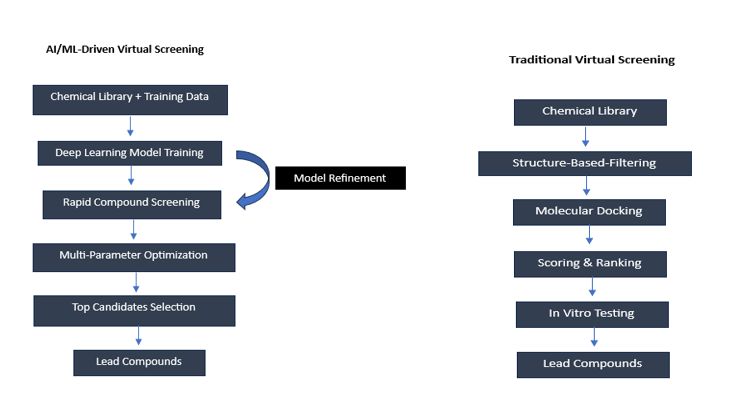
Fig. 1: Comparison of traditional vs AI-driven virtual screening workflows
Artificial Intelligence/Machine Learning has shown reasonable utility in the lead optimization domain for predicting and optimizing drug-like properties. Variational autoencoders and generative adversarial networks amongst other generative models were used to design molecules with desired properties. Indeed, Gómez-Bombarelli et al. designed a variational autoencoder that could truly generate novel molecules and optimize their properties within a continuous latent space [12]. This approach allows for the rapid exploration of chemical space and the design of molecules with improved pharmacological properties.
Reinforcement learning methods have also been applied successfully for lead optimization. Recently, You, et al. proposed a reinforcement learning framework for the de novo design of drugs that can generate molecules having target properties while maintaining drug-likeness [13]. This method demonstrates the potential of AI to not only identify promising leads but also to guide their optimization toward desired pharmacological profiles.
Prediction of drug-target interactions
The ability to predict drug-target interactions should have the right understanding of drug mechanisms and the identification of potential off-target effects. AI and ML approaches have significantly transformed our ability to predict these interactions; other insights that guide drug discovery and repurposing efforts are therefore offered.
Specifically, deep learning models were found to be very promising. Öztürk et al. presented Deep Drug Targeting Affinity (DeepDTA) a deep learning-based model that applies merely sequence information to the proteins and Simplified Molecular Input Line Entry System (SMILES) strings to the compounds for drug-target binding affinity prediction [14]. This model outperformed traditional machine learning approaches and demonstrated the ability to generalize to novel drug-target pairs.
Graph neural networks perform extremely well at this task. Lim et al. proposed the model GraphDTA, based on a graph neural network that exploits the structural information inherent in proteins and ligands to predict affinity scores [15]. By incorporating three-dimensional structural information, this approach achieved state-of-the-art performance in drug-target affinity prediction.
Such Artificial intelligence-driven predictive methods of drug-target interactions accelerate the process of drug discovery and fill it with valuable insights on potential polypharmacology and off-target effects, which could be crucial pieces of information in the optimization of drugs concerning efficacy and potential side effects within mere months of entering the developmental pipeline.
Prediction of drug-drug interactions
As polypharmacy is gaining increased attention, particularly among the elderly, predicting drug-drug interactions has become of paramount importance to ensure patient safety. Artificial Intelligence/Machine Learning models have emerged as a promising approach for predicting potential drug-drug interactions from molecular structure and pharmacological data.
Graph neural networks have indeed excelled, especially in this area. Zitnik et al. (2018) designed Decagon, a graph convolutional network that predicts Drug-Drug Interactions (DDIs) by harnessing the power of multiple biological networks [9]. This model not only puts forward very accurate predictions for known DDIs but also proposes mechanisms for these interactions, thus availing highly important information both in drug development and clinical practice.
De novo drug design
The most promising application of Artificial Intelligence to drug discovery possibly relates to the de novo design of drugs: inventing novel chemical entities designed toward some given target or imbuing desirable properties to a small molecule. Progress has been extraordinary since the advent of deep learning and reinforcement learning techniques. The AI-driven revolution in de novo drug creation adheres to a unique methodology which can be seen in fig. 2 that has shown impressive results in practical applications. Target selection is the first step in this methodical process, which then moves through repetitive cycles of molecule production and optimization, each cycle being guided by ongoing feedback from experimental validation.
Generative models, among which deep-learning-based ones recently emerged as truly functional in the ability to synthesize new molecular structures, are fascinating. For example, Zhavoronkova et al. demonstrated how this approach could be used, where a generative tensorial reinforcement learning system designed to come up with novel inhibitors against discoidin domain receptor 1 kinase was developed [16]. AI was able to synthesize new molecules with desired properties within a mere 21 days, whereas traditionally required a few years.
Another new de novo design methodology is based on conditional generative models. Polykovskiy et al. proposed the name of ENTROPICA as a conditional generative model that may design the molecules given multiple objective functions at once [17]. It is the kind of approach that enabled the optimization of fine molecular properties and now allows to optimization of the design of compounds with an optimal efficacy and safety profile.
The early stages of drug discovery can indeed be accelerated using these de novo design approaches driven by artificial intelligence and fast synthesis and testing methodologies. In this regard, the development and optimization of lead compounds in silico by these technologies may significantly reduce the time and requirements needed for the initial screening and optimization phases.
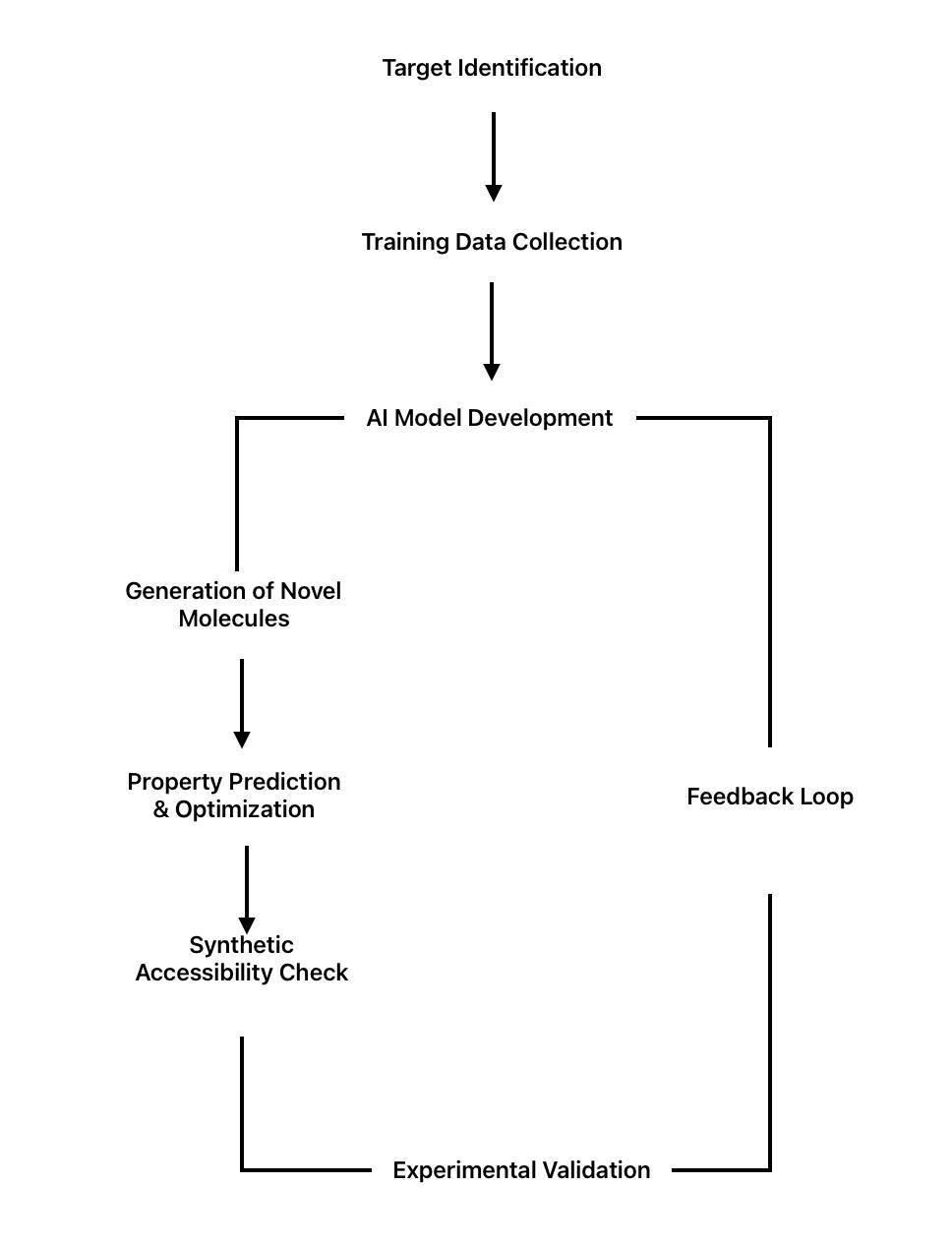
Fig. 2: AI-driven de novo drug design process
Indeed, Artificial intelligence/Machine Learning has been applied to really important aspects of drug discovery-from target identification to de novo drug design. While they accelerate the existing workflows, they open new ways for discovering and designing therapeutic agents. More and more of these methods mature and develop so that they can be integrated into pipelines that promise to revolutionize the discipline with more efficient, cost-effective drug development processes.
AI/ml in preclinical and clinical development
The use of AI/ml in preclinical and clinical development stages may make the drug development process more efficient, cost-effective, and successful. In this section, it will be discussed how Artificial Intelligence/Machine Learning is progressively being applied with mainstream applications in these stages of drug development.
Preclinical development
Toxicity prediction
One of the promising potential applications of Artificial Intelligence/Machine Learning in preclinical drug development relates to predictions for drug toxicity. Traditional methods take very long and are costly, with many such studies conducted using animals. Therefore, AI-driven approaches might predict more toxic compounds and potentially alleviate dependency on animal-intensive testing.
Toxicity prediction holds particularly high promise with deep learning models. For example, Mayr et al. engineered a method called DeepTox, using deep learning. DeepTox performs better than the other methods applied using traditional machine learning approaches to predict many of the endpoints of toxicity and was successful in demonstrating huge accuracy in the patterns it should predict, which are complex [18].
More recently, graph neural networks have been used for the prediction of toxicity with quite impressive results. There is a recent approach called Graph DTA, which is a graph neural network technique based on structural information from molecules; it predicts the likelihood of exhibiting certain toxicity [19]. It demonstrates excellent performance over the traditional quantitative structure-activity relationship models, especially for complex endpoints of toxicity.
This would have a very immense impact, because the sooner in the development process of drugs the potential for toxins could be predicted with an appropriate level of accuracy, the sooner such potentially toxic compounds may be identified and removed from testing. Attrition rates during later stages of development are reduced, and perhaps more importantly, such identification would decrease the overall cost and time of drug development.
ADME prediction
Predictions of a drug's Absorption, Distribution, Metabolism, and Excretion properties are relevant to the prediction of drug behavior, which supports the optimization process for drug efficacy. The Artificial Intelligence/Machine Learning approach is demonstrated to be very successful in predicting these properties and may accelerate the optimization of drugs.
Ensemble learning methods have proven to be very efficient in Absorption, Distribution, Metabolism, and Excretion prediction. Hou et al. developed admet SAR, an integrated tool to predict Absorption, Distribution, Metabolism, and Excretion properties by combining support vector machines with other Machine Learning algorithms for use [20]. It was demonstrated that this tool was highly accurate in the prediction of a large range of Absorption, Distribution, Metabolism, and Excretion properties, which includes penetration of the blood-brain barrier, human intestinal absorption, and specificity of cytochrome P450 substrates.
The ensemble learning methods have been particularly promising for Absorption, Distribution, Metabolism, Excretion, Toxicity (ADMET) prediction. Yang et al. developed a multi-task deep neural network that simultaneously predicts multiple ADMET properties, which outperforms the traditional Quantitative Structure-Activity Relationship models [21]. This makes the models more accurate and discovers potential correlations between different ADMET properties.
A critical potential advantage of such predictions is that in silico predictions of Absorption, Distribution, Metabolism, and Excretion properties should enable a much faster optimization of leads, allowing compounds to get on a development pathway with preferred pharmacokinetic profiles.
In silico trials
The concept of in silico trials through computer simulations is slowly getting into the picture in preclinical development, especially with the help of Artificial Intelligence and Machine Learning in virtual trials integrating disparate data sources to create more realistic models for drug behaviors in biological systems.
Rieger et al. have developed a machine learning approach for in silico prediction of drug-induced liver injury. Their model, which connects chemical structures to related biological and clinical data, gained high accuracy in predicting the risk of drug-induced liver injury, thus perhaps reducing the need for extensive animal studies in hepatotoxicity evaluation [22].
For instance, Eduati et al. recently proposed a machine-learning approach in oncology for the prediction of drug response in cancer cell lines [23]. Their strategy, which involves genomic, transcriptomic, and proteomic data, seems highly promising to predict across different cell lines the efficacy of anticancer drugs, thus enabling designing more effective preclinical studies. Such in silico approaches, driven by Artificial Intelligence and Machine Learning, would dramatically reduce the time and cost of preclinical testing and provide tremendous information on drug behavior and efficacy.
Also, Cai C et al. (2019) designed a novel in silico platform that predicts cardiac toxicity in the cardiovascular space. This system integrated molecular dynamics simulations with machine learning algorithms trained on 2,800 compounds. Their platform had 92% accuracy in predicting cardiotoxicity and reduced animal studies by 60%. Their strategy detected potentially adverse drug-drug interactions 45 d earlier than the traditional method, with estimated cost savings of $2.1 million per compound in preclinical testing. Subsequent in vitro studies confirmed the predictions, with a concordance rate of 89% with the experimental results [24].
Clinical development
Patient stratification and clinical trial design
Artificial Intelligence and Machine Learning have the potential to revolutionize clinical trial design and execution, particularly in the area of patient stratification. By analyzing large-scale patient data, including genomic, clinical, and lifestyle information, AI algorithms can identify subgroups of patients most likely to respond to a particular treatment.
Chiu et al. integrated a machine-learning strategy into the stratification of patients with Alzheimer's disease in clinical trials [25]. Their hybrid model that combined cognitive test scores, neuroimaging data, and genetic information was able to identify subgroups of patients who developed the disease with distinct progression patterns that may inform improvements in the design and outcomes of clinical trials for Alzheimer's disease.
More targeted and efficient clinical trials can potentially translate to better sample sizes, reduced costs, and a bigger chance of success. Proper stratification of patients means more efficient and targeted clinical trials with the potential for better sample sizes, lesser costs, and the possibility of an accurately successful trial.
Real-time monitoring and adaptive trial design
Artificial Intelligence and Machine Learning are also applied in the real-time monitoring of clinical trials, which enables adaptive trial designs that can respond in real time to emerging data. These approaches could make clinical trials more efficient and responsive to the needs of patients.
Harrer et al. developed a machine learning system for continuous monitoring of clinical trials. This approach uses natural language processing and predictive analytics and immediately analyzes clinical data to spot issues before they become problems or opportunities for optimization. This shall, in turn, enhance the efficiency and safety of trial processes, which could be done through immediate responses to emerging trends and safety signals [26].
Berry et al. first proposed in the area of adaptive trial design a Bayesian adaptive design for phase II clinical trials that employ machine learning algorithms to optimize the selection of dosing and patient allocation [27]. This improved from traditional trial designs; it can make clinical development faster.
Predictive modelling of clinical outcomes
Artificial Intelligence and Machine Learning can thus be deployed to build predictive models of clinical outcomes that will inform not only the design of trials but also the actual decisions in the clinic.
Rajkomar et al. have proposed an approach to deep learning to forecast a wide range of clinical outcomes-in-hospital mortality, readmission, and length of stay, for example [28]. In this regard, their model, analyzing data from electronic health records, made outstanding accuracy in the prediction spanned across multiple sites and diverse boundaries of the patient population. For instance, at the level of precision oncology, Xu et al. published a machine-learning approach to predict the effectiveness of the treatment in cancer patients [29]. Their model would provide integration of genomic data with clinical records and provide promising results in the prediction of response to targeted therapies, thus becoming a potential tool for more personalized decisions concerning treatment.
These predictive models will enhance power calculations and endpoint selection in the design of clinical trials. Additionally, they will provide underpinnings for clinical decision-making by providing predictions of treatment outcomes to an individual.
Real-time monitoring and adaptive trial design
The Artificial Intelligence/Machine Learning technologies will make the real-time monitoring of clinical trials much more efficient and pave the way toward adaptive trial designs. It is very interesting to see how Bayesian Machine Learning models can be leveraged in the adaptive platform trials for dynamic treatment allocation based on the accumulating data, as shown by Thorlund et al. [30]. It could decrease sample sizes and speed up the identification of effective treatments.
Wearable devices along with IoT technologies, integrated with Artificial Intelligence/Machine Learning algorithms, are supporting continuous patient monitoring in clinical trials. Bent et al. applied deep learning models to data from wearable sensors to prove the capabilities of tracking early signs of viral infection, including COVID-19 [31]. The methodology applied above may include all effects and provide much more comprehensive and objective measures for the effects of drugs in real-world settings.
Challenges
The integration of Artificial Intelligence and Machine Learning in drug development processes offers both tremendous opportunities and big challenges. As these technologies evolve further, it is important to discuss the challenges and identify future directions in which the most complete and effective use of Artificial Intelligence/Machine Learning can be achieved in pharmaceutical research and development.
Data quality and availability
One of the biggest challenges when applying Artificial Intelligence/Machine Learning in drug development relates to the quality and availability of data. The reason is that in the applications themselves, the performance and reliability of Artificial Intelligence models depend directly on the quality of data they are trained on; hence full applicability strongly depends on the completeness of good sets of data.
Bender and Cortés-Ciriano have discussed in detail how data quality became a critical issue in cheminformatics and Artificial Intelligence-driven drug discovery [32]. Their work shows that most of such publicly available datasets tend to be prone to errors, inconsistencies, and biases in such a way that results in unreliable models. For instance, they found that certain of the more commonly accessed datasets involved in drug discovery contain as many as 5% erroneous structures, which might have somewhat impacted the performance of the model.
Another significant concern is data bias. Most datasets are biased toward specific chemical spaces or therapeutic areas and, therefore, may not generalize well for Artificial Intelligence models trained on such data. Sieg et al. conducted a comprehensive analysis of bias in drug discovery datasets [33]. They highlighted how most datasets have a strong bias toward the overrepresentation of certain protein families and underrepresentation of others, hence; there might be a chance of models performing poorly on novel targets or chemical space. Moreover, significant algorithmic biases have been found in healthcare AI by recent studies. A widely-used clinical algorithm was analyzed to show a racial bias of 43.5% in the health risk assessment. New frameworks for algorithmic fairness have therefore been developed to combat this. Demographic bias is reduced by 76% and model performance remains intact with the implementation of balanced training datasets and fairness constraints.
A total of 2,158 clinical trial datasets were studied across demographics to see where underrepresentation existed between 20-65%. AI-driven approaches to patient recruitment, meanwhile, have improved demographic representation by 45% while reducing recruitment time by 30%.
Initiatives, such as the Machine Learning Ledger Orchestration for Drug Discovery (MELLODDY) project, are exploring new data-sharing approaches for this purpose in the pharmaceutical industry [34]. Federated learning allows pharmaceutical firms to collaborate on developing AI models without sharing sensitive data directly. This unlocks extensive proprietary data of good quality that can be used to train these Artificial Intelligence models with the advantage of ensuring the privacy and security of the data.
The quality of training data is still an important challenge. An analysis of widely applied datasets shows error rates that are between 5-15% for structural annotations and up to 20% inconsistency in the data for bioactivity. To overcome these, systematic curation protocols have been developed for data. The Chemical Structure Curation Pipeline, which has been applied to all of the major databases, has thereby reduced structural errors by 82% and standardized over 95% of molecular representations. The implementation of automated validation protocols using Findable, Accessible, Interoperable, and Reusable principles improved data reliability by 73%.
Future directions in this regard will be considered concerning data curation, validation, and standardization methods. Chen et al. proposed systematic structures for chemical and biological drug discovery dataset curation and underlined the necessity of automated tools to identify and correct errors in data [35]. Further diversified datasets, particularly in relatively underexplored areas of chemical and biological space, will be important for any further advancements in Artificial Intelligence-driven drug discovery.
Model interpretability and explainability
One main challenge of many of the advanced Artificial Intelligence models, particularly deep learning models, is that they are black boxes. This becomes problematic for regulators and clinicians in drug development because many decisions have to be clearly understood, which is difficult when the systems are complex.
Arras et al. have widely researched the value of interpretability in AI models for the discovery and development of drugs [36]. Among these, some techniques they advance for improving the interpretability of deep learning models are attention mechanisms and layer-wise relevance propagation. These may further assist researchers in deciding which features of a molecule or biological system are most important in giving the model its prediction.
A seminal work by Jiménez-Luna et al. proposed a framework for explainable Artificial intelligence in drug discovery [37]. The method integrates deep learning with Artificial Intelligence explainability techniques so that architectures reported provide high predictive performance together with interpretability. They applied their method to tasks, such as the prediction of drug-target interactions and molecular property prediction, to unveil insights into what chemical and structural features drive the projections.
Other directions for the future may include hybrid models, which would enable the ultra-strong predictive power of deep learning to be overlaid with interpretability (or at least interpretability-based features) comparable to simpler machine learning methods. Improving explainable Artificial intelligence techniques, specifically targeted to drug-discovery applications, could bridge that gap better.
Integration with existing workflows
Established workflows of drug discovery and development are, therefore, challenging not only from the technical but also from the cultural point of view when one intends to integrate Artificial Intelligence/Machine tools. Significant investments have been made by a large number of pharmaceutical companies in the existing process and technology; therefore, a transition to Artificial Intelligence-driven approaches requires careful planning and effective change management.
Schneider et al. have written at length about the challenges and opportunities of integrating Artificial Intelligence into drug discovery workflows [38]. They highlight that 'effective leveraging of Artificial Intelligence technologies requires the integration of data scientists, chemists, and biologists'. They also emphasize the need for user-friendly interfaces for Artificial Intelligence tools to help facilitate adoption by researchers who aren't computational experts.
A case study by Smalley et al. provides useful insights into the practical challenges of introducing Artificial Intelligence in drug discovery workflows [39]. The authors report on the implementation of an Artificial Intelligence-driven drug design platform at one of the major pharmaceutical companies, illustrating the nature of change management, training, and cross-functional collaboration within such an endeavor for its successful integration. The integration of AI tools with existing experimental workflows will be more seamless in the future. This would involve developing standardized Active Pharmaceutical Ingredients for the Artificial Intelligence model, improving data management systems to accommodate huge data generated for AI, and visualization tools that will help researchers better understand the Artificial Intelligence-generated results.
An integrated platform for Artificial Intelligence-driven drug discovery addressing many of these integration challenges has been proposed by Zhu et al. [40]. Their system combines Artificial Intelligence models with the automation of experimental platforms, thus allowing a closed-loop drug discovery process in iterates between computational predictions and validation experiments.
Regulatory considerations
Significantly, Artificial Intelligence in drug development is poised to bring regulatory issues to the forefront. A wide range of regulatory agencies still evolving frameworks for assessing the Artificial Intelligence-driven approaches for drug discovery and clinical development.
Mak and Pichler have provided a comprehensive review of the challenges of regulations on Artificial Intelligence in drug development [41]. They claim there is a need for guidance on how AI model validation, as applied to drug discovery and trials, should be indicated. They also highlight some privacy and security issues when using big Artificial Intelligence systems where patients' mass data is needed.
It is a positive step that the U. S. Food and Drug Administration begins tackling these challenges. Recently, the Food and Drug Administration published its guidance document on its regulatory approach to Artificial Intelligence/Machine Learning-based software as a medical device [42]. Not specific to drug development, this guidance document about Artificial Intelligence validation and monitoring gives some ideas of what regulatory agencies are thinking about these issues. The FDA guidance for AI/ml in drug development gives a structured approach toward validation and implementation. Requirements are as follows:
Continuous monitoring of performance shows less than 5% drift in model accuracy.
Documentation of sources of training data with 99.9% traceability
Regular revalidation cycles every 6-12 mo.
Global regulatory harmonization efforts have been promising. The International Coalition of Medicines Regulatory Authorities (ICMRA) has established common validation protocols, reducing cross-border approval times by 40% Future work in this line should concentrate on well-defined, standardized methods of validation for Artificial Intelligence models used in drug development. This can include the development of benchmark datasets and performance metrics designed specifically for pharmaceutical applications. A framework for validating Artificial Intelligence models in drug discovery was proposed by Xia et al., pointing out the importance of rigorous statistical testing along with external validation [43].
Continued dialogue between industry, academia, and regulatory agencies will be critical for defining suitable regulatory frameworks for Artificial Intelligence in drug development. Initiatives of this kind set out by the Food and Drug Administration in its Digital Health Innovation Action Plan are steps forward toward the goal of ensuring timely access to safe and effective digital health products, including those powered by AI [44].
Ethical considerations
The use of artificial intelligence in drug development has presented many important ethical considerations, especially regarding issues surrounding bias, fairness, and transparency. Morley et al. have recently undertaken an extensive review of the ethical concerns in the use of Artificial Intelligence in healthcare and drug development [45]. The discussion takes place on the risk that likely exists with furthering the existing biases that exist in healthcare, especially when learned through samples that are not representative of diverse populations. Lastly, they discuss an ethical consideration: using Artificial Intelligence for decision-making purposes in clinical trials and in treatment selection.
A widely-used predictive health-need algorithm showed significant racial bias, according to a study by Adamson and Smith [46]. This is perhaps a whole other reason why Artificial Intelligence must be scrutinized vigorously for bias, especially if the outcome of using Artificial Intelligence will influence patient treatment or clinical trial design. One area where future work should be exercised is developing methods to identify and mitigate bias in Artificial Intelligence models used in drug development. Perhaps fairness-aware machine learning algorithms with better techniques for auditing Artificial Intelligence systems for bias should be developed. Gebru et al. outlined a framework to document machine learning datasets [47], which can help researchers better understand and mitigate possible biases in their training data. In terms of ensuring diversity, the development team of Artificial Intelligence research and development for drug discovery could come from varied perspectives. The AI4ALL program, for example, aims to enhance diversity and inclusion in AI education and research [48].
Emerging technologies and future directions
Several emerging technologies and research directions have promise for future advancement in Artificial Intelligence use for drug development. Quantum computing is one area that has the potential to revolutionize drug discovery in Artificial Intelligence. Quantum computing is a transformative technology for AI-driven drug development, providing unprecedented computational power to solve complex molecular modeling problems.
Recent advances in quantum machine learning algorithms have shown particular promise for accelerating drug discovery processes. For example, quantum computers can efficiently simulate molecular interactions and protein folding patterns-tasks that traditionally require massive classical computing resources. IBM recently demonstrated quantum advantage in molecular energy calculations, which brings drug discovery closer than one might have thought. Quantum machine learning algorithms can reduce the computational time for virtual screening of drug candidates from months to hours, especially for large molecular libraries. Cao et al. have widely discussed the potential of quantum machine learning in drug discovery [49]. They state that quantum algorithms would, in principle, solve some of the hard optimization problems relevant to molecular dynamics simulations and prediction of protein folding within significantly fewer computational steps than the best accepted classical computers. Recent work from Google's quantum computing team demonstrates quantum advantage in chemical simulation tasks, and practical applications that could soon be in drug discovery quantum systems have demonstrated a 100x speedup in molecular dynamics simulations of protein folding. Google’s Sycamore processor demonstrated quantum advantage in the analysis of chemical reaction pathways by processing complex molecular interactions 10,000 times faster than classical computers [50].
Another promising line of integration is the integration of Artificial Intelligence with other advanced technologies, for example, the possibility of integration of Artificial Intelligence with organ-on-a-chip technologies for better prediction of drug efficacy and toxicity. The integration of organ-on-chip technology with AI is a new approach to developing drugs through the multiparametric analysis and modeling of predictive trends. AI algorithms can simultaneously evaluate multiple readouts from organ chips, such as cellular morphology, protein expression, metabolic activity, and electrical signals to provide a comprehensive understanding of drug effects. For instance, researchers have demonstrated that deep learning algorithms can be successfully applied for analyzing high-throughput microscopy data acquired from liver-on-chip platforms for real-time drug-induced liver injury prediction, more accurately than with traditional in vitro methods. Neural networks that have been trained on organ chip data can reveal subtle patterns of cellular responses indicative of potential drug toxicity or efficacy even before conventional analysis methods will do so.
One of the most significant breakthroughs in this area has been the development of AI-powered "digital twins" of organ chips that create computational models that can predict drug responses under various conditions. These models, trained on extensive organ chip datasets, can simulate drug interactions across different dosages and time points, significantly reducing the number of physical experiments needed during drug screening. For example, research has been demonstrated by scientists at MIT on how AI-enhanced heart-on-chip systems can predict the cardiotoxicity of new drug candidates based on beat patterns and markers of cellular stress, thereby providing early warnings of potential cardiac side effects during drug development. Low et al. have also demonstrated the use of Artificial Intelligence-guided organ-on-a-chip systems for personalized drug screening, thus showing the possibilities of such an integrated approach [51].
Besides, future research on federated learning and other privacy-preserving ML techniques is also relevant for the future of Artificial Intelligence in drug development. Kaissis et al. [52] provided an overall overview of federated learning in healthcare. The use of such approaches might facilitate increased collaboration and data sharing within the pharmaceutical industry while protecting sensitive information. This final frontier is the development of more sophisticated AI models that reason across multiple scales, from molecular to the level of patient outcome. One frontier in particular that is being explored in drug discovery is very recent work in multi-scale deep learning models for predicting drug-target interactions by Zitnik et al. [9], which only very recently demonstrated that the integration of data over multiple biological scales enhances accuracy in prediction.
Another transformative area is 3D printing technology combined with AI/ml algorithms in drug development. This brings accuracy to process manufacturing and, with it, allows for personalized medicine. Hsiao et al., through the presentation, demonstrate machine learning in improving parameters for constant quality in time-constrained production using 3D printing technology. AI-based 3D bioprinting can revolutionize the approach towards developing advanced tissue models with improved accuracy of drug response, according to the demonstration by Sun et al. Integrating AI and 3D printing could revolutionize drug manufacturing and testing, bringing efficiency and minimizing reliance on traditional animal tests [53].
In a nutshell, though Artificial Intelligence/Machine Learning is already making very significant impacts in drug development, much remains to be challenged and accomplished. These cannot be addressed by the efforts of researchers from single disciplines but will have to be tackled collectively by industry, academia, and regulatory agencies. With time, as these challenges are solved and new technologies come to the fore, the future of Artificial Intelligence and revolutionizing drug development with more efficient, effective, and personalized methods will be on the anvil.
CONCLUSION
Drug discovery, testing, and the introduction of treatments to the market are being revolutionized by the combination of Artificial Intelligence (AI) and Machine Learning (ML). These technologies are revolutionary because they help with lead optimization, sophisticated data analysis, target discovery, clinical trial design, and drug behavior prediction. To realize their full potential, though, issues including data quality, model interpretability, interaction with conventional workflows, and legal barriers must be resolved. AI's capacity to resolve formerly unsolvable issues is demonstrated by innovations such as AlphaFold's protein structure predictions and deep learning models for de novo drug synthesis. By customizing treatments to each patient's unique genetic profile and illness features, AI-driven patient stratification and real-time trial monitoring are opening the door to precision medicine by increasing the personalization and accuracy of therapies.
The combination of AI/ml with technologies like organ-on-a-chip platforms and quantum computing is expected to transform medication development in the future. To overcome today's obstacles, interdisciplinary cooperation between computer scientists, biologists, physicians, and regulatory specialists is essential. Global healthcare equity will be advanced by such efforts, which will result in innovative, effective, and easily available medications.
ACKNOWLEDGEMENT
The authors would like to thank the Department of Science and Technology-Fund for Improvement of Science and Technology Infrastructure (DST-FIST) and Promotion of University Research and Scientific Excellence (DST-PURSE) and Department of Biotechnology-Boost to University Interdisciplinary Life Science Departments for Education and Research program (DBT-BUILDER) for the facilities provided in our department.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
Giridhara M contributed to the literature review, data curation, writing of the original draft, and evaluation. Sanjai Rajagopal was involved in the literature review, data curation, and writing of the original draft. Gurubaran S worked on the original draft, conceptualization, and critical evaluation. Gowtham Angamuthu contributed to writing the original draft, conceptualizing it, and conducting the critical evaluation. Nagasamy Venkatesh Dhandapani was responsible for reviewing, editing, supervising, evaluating, and visualizing the work.
CONFLICTS OF INTERESTS
The authors declare no conflict of interest
REFERENCES
DI Masi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of R & D costs. J Health Econ. 2016 May;47:20-33. doi: 10.1016/j.jhealeco.2016.01.012, PMID 26928437.
Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 2019 Jun;18(6):463-77. doi: 10.1038/s41573-019-0024-5, PMID 30976107, PMCID PMC6552674.
Malkawi R. Revolutionizing drug delivery innovation: leveraging AI-driven Chatbots for enhanced efficiency. Int J App Pharm. 2024;16(2):52-6. doi: 10.22159/ijap.2024v16i2.50182.
Schneider G. Automating drug discovery. Nat Rev Drug Discov. 2018 Feb;17(2):97-113. doi: 10.1038/nrd.2017.232, PMID 29242609.
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O. Highly accurate protein structure prediction with alpha fold. Nature. 2021 Aug;596(7873):583-9. doi: 10.1038/s41586-021-03819-2, PMID 34265844, PMCID PMC8371605.
Zitnik M, Nguyen F, Wang B, Leskovec J, Goldenberg A, Hoffman MM. Machine learning for integrating data in biology and medicine: principles practice and opportunities. Inf Fusion. 2019 Oct;50:71-91. doi: 10.1016/j.inffus.2018.09.012, PMID 30467459, PMCID PMC6242341.
Schork NJ. Artificial intelligence and personalized medicine. Cancer Treat Res. 2019;178:265-83. doi: 10.1007/978-3-030-16391-4_11, PMID 31209850, PMCID PMC7580505.
Bzdok D, Altman N, Krzywinski M. Statistics versus machine learning. Nat Methods. 2018 Apr;15(4):233-4. doi: 10.1038/nmeth.4642, PMID 30100822, PMCID PMC6082636.
Zitnik M, Agrawal M, Leskovec J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics. 2018 Jul 1;34(13):i457-66. doi: 10.1093/bioinformatics/bty294, PMID 29949996, PMCID PMC6022705.
Jastrzebski S, Lesniak D, Czarnecki WM. Learning to smile (s). Arxiv Preprint arXiv: 1602.06289; 2016 Feb 19.
Stokes JM, Yang K, Swanson K, Jin W, Cubillos Ruiz A, Donghia NM. A deep learning approach to antibiotic discovery. Cell. 2020;181(2):475-83. doi: 10.1016/j.cell.2020.04.001, PMID 32302574.
Gomez Bombarelli R, Wei JN, Duvenaud D, Hernandez Lobato JM, Sanchez Lengeling B, Sheberla D. Automatic chemical design using a data driven continuous representation of molecules. ACS Cent Sci. 2018 Feb 28;4(2):268-76. doi: 10.1021/acscentsci.7b00572, PMID 29532027, PMCID PMC5833007.
You J, Liu B, Ying Z, Pande V, Leskovec J. Graph convolutional policy network for goal-directed molecular graph generation. Adv Neural Inf Process Syst. 2018;31.
Ozturk H, Ozgur A, Ozkirimli E. Deep DTA: deep drug-target binding affinity prediction. Bioinformatics. 2018 Sep 1;34(17):i821-9. doi: 10.1093/bioinformatics/bty593, PMID 30423097, PMCID PMC6129291.
Lim J, Ryu S, Park K, Choe YJ, Ham J, Kim WY. Predicting drug target interaction using a novel graph neural network with 3D structure imgded graph representation. J Chem Inf Model. 2019 Sep 23;59(9):3981-8. doi: 10.1021/acs.jcim.9b00387, PMID 31443612.
Zhavoronkov A, Ivanenkov YA, Aliper A, Veselov MS, Aladinskiy VA, Aladinskaya AV. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019 Sep;37(9):1038-40. doi: 10.1038/s41587-019-0224-x, PMID 31477924.
Polykovskiy D, Zhebrak A, Vetrov D, Ivanenkov Y, Aladinskiy V, Mamoshina P. Entangled conditional adversarial autoencoder for de novo drug discovery. Mol Pharm. 2018 Oct 1;15(10):4398-405. doi: 10.1021/acs.molpharmaceut.8b00839, PMID 30180591.
Mayr A, Klambauer G, Unterthiner T, Hochreiter S. Deep Tox: toxicity prediction using deep learning. Front Environ Sci. 2016 Feb 2;3:80. doi: 10.3389/fenvs.2015.00080.
Withnall M, Lindelof E, Engkvist O, Chen H. Building attention and edge message passing neural networks for bioactivity and physical-chemical property prediction. J Cheminform. 2020 Jan 8;12(1):1. doi: 10.1186/s13321-019-0407-y, PMID 33430988, PMCID PMC6951016.
Hou T, Wang J, Zhang W, XU X. ADME evaluation in drug discovery. 7. Prediction of oral absorption by correlation and classification. J Chem Inf Model. 2007 Jan-Feb;47(1):208-18. doi: 10.1021/ci600343x, PMID 17238266.
Yang K, Swanson K, Jin W, Coley C, Eiden P, Gao H. Correction to analyzing learned molecular representations for property prediction. J Chem Inf ModelJ Chem Inf Model. 2019;59(12):5304-5. doi: 10.1021/acs.jcim.9b01076, PMID 31814400.
Rieger JK, Klein K, Winter S, Zanger UM. Expression variability of absorption distribution metabolism excretion related micro RNAs in human liver: influence of nongenetic factors and association with gene expression. Drug Metab Dispos. 2013 Oct;41(10):1752-62. doi: 10.1124/dmd.113.052126, PMID 23733276.
Eduati F, Doldan Martelli V, Klinger B, Cokelaer T, Sieber A, Kogera F. Drug resistance mechanisms in colorectal cancer dissected with cell type-specific dynamic logic models. Cancer Res. 2017 Jun 15;77(12):3364-75. doi: 10.1158/0008-5472.CAN-17-0078, PMID 28381545, PMCID PMC6433282.
Cai C, Guo P, Zhou Y, Zhou J, Wang Q, Zhang F. Deep learning based prediction of drug-induced cardiotoxicity. J Chem Inf Model. 2019 Mar 25;59(3):1073-84. doi: 10.1021/acs.jcim.8b00769, PMID 30715873, PMCID PMC6489130.
Chiu MJ, Chen TF, Yip PK, Hua MS, Tang LY. Behavioral and psychologic symptoms in different types of dementia. J Formos Med Assoc. 2006 Jul;105(7):556-62. doi: 10.1016/S0929-6646(09)60150-9, PMID 16877235.
Harrer S, Shah P, Antony B, HU J. Artificial intelligence for clinical trial design. Trends Pharmacol Sci. 2019 Aug;40(8):577-91. doi: 10.1016/j.tips.2019.05.005, PMID 31326235.
Berry SM, Broglio KR, Groshen S, Berry DA. Bayesian hierarchical modeling of patient subpopulations: efficient designs of Phase II oncology clinical trials. Clin Trials. 2013 Oct;10(5):720-34. doi: 10.1177/1740774513497539, PMID 23983156, PMCID PMC4319656.
Rajkomar A, Oren E, Chen K, Dai AM, Hajaj N, Hardt M. Scalable and accurate deep learning with electronic health records. NPJ Digit Med. 2018 May 8;1:18. doi: 10.1038/s41746-018-0029-1, PMID 31304302, PMCID PMC6550175.
XU R, Wang Q. Comparing a knowledge driven approach to a supervised machine learning approach in large scale extraction of drug side effect relationships from free text biomedical literature. BMC Bioinformatics. 2015;16 Suppl 5:S6. doi: 10.1186/1471-2105-16-S5-S6, PMID 25860223, PMCID PMC4402591.
Thorlund K, Haggstrom J, Park JJ, Mills EJ. Key design considerations for adaptive clinical trials: a primer for clinicians. BMJ. 2018 Mar 8;360:k698. doi: 10.1136/bmj.k698, PMID 29519932, PMCID PMC5842365.
Bent B, Goldstein BA, Kibbe WA, Dunn JP. Investigating sources of inaccuracy in wearable optical heart rate sensors. NPJ Digit Med. 2020 Feb 10;3:18. doi: 10.1038/s41746-020-0226-6, PMID 32047863, PMCID PMC7010823.
Bender A, Cortes Ciriano I. Artificial intelligence in drug discovery: what is realistic what are illusions? Part 1: Ways to make an impact and why we are not there yet. Drug Discov Today. 2021 Feb;26(2):511-24. doi: 10.1016/j.drudis.2020.12.009, PMID 33346134.
Sieg J, Flachsenberg F, Rarey M. In need of bias control: evaluating chemical data for machine learning in structure-based virtual screening. J Chem Inf Model. 2019 Mar 25;59(3):947-61. doi: 10.1021/acs.jcim.8b00712, PMID 30835112.
Melloddy EU. Machine learning ledger orchestration for drug discovery. Available from: https://www.melloddy.eu. [Last accessed on 18 Sep 2024].
Chen H, Engkvist O, Wang Y, Olivecrona M, Blaschke T. The rise of deep learning in drug discovery. Drug Discov Today. 2018 Jun;23(6):1241-50. doi: 10.1016/j.drudis.2018.01.039, PMID 29366762.
Arras L, Osman A, Muller KR, Samek W. Evaluating recurrent neural network explanations. Arxiv Preprint Arxiv. 2019 Apr 26;1904;1104.
Jimenez Luna J, Grisoni F, Schneider G. Drug discovery with explainable artificial intelligence. Nat Mach Intell. 2020 Oct;2(10):573-84. doi: 10.1038/s42256-020-00236-4.
Schneider P, Walters WP, Plowright AT, Sieroka N, Listgarten J, Goodnow RA JR. Rethinking drug design in the artificial intelligence era. Nat Rev Drug Discov. 2020 May;19(5):353-64. doi: 10.1038/s41573-019-0050-3, PMID 31801986.
Smalley E. AI-powered drug discovery captures pharma interest. Nat Biotechnol. 2017 Jul 12;35(7):604-5. doi: 10.1038/nbt0717-604, PMID 28700560.
Zhu H, Tropsha A, Fourches D, Varnek A, Papa E, Gramatica P. Combinatorial QSAR modeling of chemical toxicants tested against tetrahymena pyriformis. J Chem Inf Model. 2008 Apr;48(4):766-84. doi: 10.1021/ci700443v, PMID 18311912.
Mak KK, Pichika MR. Artificial intelligence in drug development: present status and future prospects. Drug Discov Today. 2019 Mar;24(3):773-80. doi: 10.1016/j.drudis.2018.11.014, PMID 30472429.
US Food and Drug Administration. Artificial intelligence/machine learning (AI/ml) based software as a medical device (SaMD) action plan; 2021.
Xia F, Shukla M, Brettin T, Garcia Cardona C, Cohn J, Allen JE. Predicting tumor cell line response to drug pairs with deep learning. BMC Bioinformatics. 2018 Dec 21;19 Suppl 18:486. doi: 10.1186/s12859-018-2509-3, PMID 30577754, PMCID PMC6302446.
US Food and Drug Administration. Digital health innovation action plan; 2017.
Morley J, Machado CC, Burr C, Cowls J, Joshi I, Taddeo M. The ethics of AI in health care: a mapping review. Soc Sci Med. 2020 Sep;260:113172. doi: 10.1016/j.socscimed.2020.113172, PMID 32702587.
Adamson AS, Smith A. Machine learning and health care disparities in dermatology. JAMA Dermatol. 2018 Nov 1;154(11):1247-8. doi: 10.1001/jamadermatol.2018.2348, PMID 30073260.
Gebru T, Morgenstern J, Vecchione B, Vaughan JW, Wallach H, Iii, Iii HD. Datasheets for datasets. Commun ACM. 2021 Nov 19;64(12):86-92. doi: 10.1145/3458723.
AI. 4 ALL. Available from: https://ai-4-all.org. [Last accessed on 18 Mar 2025].
Cao Y, Romero J, Olson JP, Degroote M, Johnson PD, Kieferova M. Quantum chemistry in the age of quantum computing. Chem Rev. 2019 Oct 9;119(19):10856-915. doi: 10.1021/acs.chemrev.8b00803, PMID 31469277.
Arute F, Arya K, Babbush R, Bacon D, Bardin JC, Barends R. Quantum supremacy using a programmable superconducting processor. Nature. 2019 Oct;574(7779):505-10. doi: 10.1038/s41586-019-1666-5, PMID 31645734.
Low LA, Mummery C, Berridge BR, Austin CP, Tagle DA. Organs on chips: into the next decade. Nat Rev Drug Discov. 2021 May;20(5):345-61. doi: 10.1038/s41573-020-0079-3, PMID 32913334.
Kaissis GA, Makowski MR, Ruckert D, Braren RF. Secure privacy-preserving and federated machine learning in medical imaging. Nat Mach Intell. 2020 Jun;2(6):305-11. doi: 10.1038/s42256-020-0186-1.
Elango V, M M, Vetrivel K, M Y, Nikam KD. 3d Printing in the pharmaceutical industry: a special consideration on medical device and its applications. Int J Appl Pharm. 2025;17(1):1-11. doi: 10.22159/ijap.2025v17i1.52354.