
Int J App Pharm, Vol 17, Issue 4, 2025, 210-215Original Article
BIOEQUIVALENCE STUDY OF A 400 MG INTRAVAGINAL PROGESTERONE PESSARY USING TANDEM MASS SPECTROMETRY IN FEMALE SUBJECTS
AHMAD ABU-AWWAD1*, BASIL ARAFAT2,3, TAWFIQ ARAFATE2
1Faculty of Pharmacy, Jerash University, P. O. Box 311, Jerash-26150, Jordan. 2Jordan Center for Pharmaceutical Research, P. O. Box 950435, Amman-11105, Jordan. 3Faculty of Health, Education, Medicine and Social Care, Anglia Ruskin University, Chelmsford Campus, UK
*Corresponding author: Ahmad Abu-Awwad; *Email: a.awwad@jpu.edu.jo
Received: 09 Jan 2025, Revised and Accepted: 22 Apr 2025
ABSTRACT
Objective: This study aimed to evaluate the bioequivalence of 400 mg single pessary dose for intravaginal progesterone test product in pilot study followed by a full study compared to the reference product, Cyclogest®. The test product represents a newly developed intravaginal progesterone formulation designed to achieve bioequivalent Pharmacokinetic (PK) parameters to Cyclogest®.
Methods: The test product was initially studied in six healthy adult females following a randomized, two-way crossover design under fasted state. The results from the pilot study were further confirmed in full study of 38 subjects. Progesterone with progesterone-D9 as the internal standard, was extracted and quantified from human plasma samples using LC-MS/MS after a direct precipitation extraction procedure. The developed method was validated over a dynamic range of 0.3–25 ng/ml in accordance with US-FDA, EMEA, and ICH guidelines.
Results: All validation results were within the acceptance criteria. The geometric mean values for the main PK parameters from the pilot bioequivalence study for the test product were Cmax= 13.094 ng/ml and AUC0-t= 168.786 h. ng/ml, compared to the results of reference product (Cmax=11.920 ng/ml and AUC0-t = 154.339 h. ng/ml), which were in agreement with the corresponding results obtained from the full study for test product (Cmax= 15.175 ng/ml and AUC0-t = 202.669 h. ng/ml) compared to the results of reference product (Cmax= 13.359 ng/ml and AUC0-t = 206.423 h. ng/ml).
Conclusion: The investigated test progesterone product was bioequivalent to the reference product.
Keywords: Progesterone, Bioequivalence, LC-MS/MS, Method validation, Pilot study
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i4.53648 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Progesterone (C21H30O2, mass 314.47 g/mol; fig. 1) is an endogenous steroid natural sex hormone that is commonly produced by the adrenal cortex and the gonads, which consist of the ovaries and the testes.
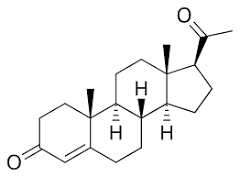
Fig. 1: Chemical structure of progesterone
Progesterone has been used for the treatment of a variety of reproductive disorders [1], and as part of hormone replacement therapy in cases of low progesterone levels [2]. Different routes of administration for progesterone are available, including oral, transdermal, intranasal, intravenous injection, intramuscular, subcutaneous, rectal, and vaginal formulations [3, 4]. The Pharmacokinetic (PK) profile of vaginal progesterone has been reported to be more than 90% of that of oral administration [5] depending on absorption rather than clearance [6]. With its easy administration and high absorption potential, the vaginal route has become the most effective way to deliver natural progesterone through the ‘uterine first-pass effect’ leading to higher tissue concentrations [7], that avoids liver first-pass metabolism, and has no systemic side-effects [8, 9]. Despite the widespread use of intravaginal progesterone, still the literature lacks to bioequivalence studies, especially those focusing on the 400 mg pessary dose. The current study was undertaken to address this gap by examining the bioequivalence of a newly developed formulation of an intravaginal progesterone in a 400 mg pessary dose, against the corresponding Cyclogest® 400 mg reference product, conducted through a pilot study, followed by a full study, where the outcome of the PK parameters obtained from the pilot study was confirmed further in the full study with 38 female subjects. Furthermore, we developed and validated a new bioanalytical method for progesterone quantification in human plasma, addressing the challenges of endogenous variability in female subjects. The current described method combines the advantages of sample preparation involving single extraction step by protein direct precipitation, and a novel Lower Limit Of Quantitation (LLOQ) of 0.3 ng/ml, followed by LC-MS/MS analysis with a high throughput total runtime of 1.6 min, selective and accurate measurements, whereas the recently reported methodologies that investigated progesterone used HPLC involving liquid-liquid extraction [10], and LC-MS/MS over the LLOQ of 1 ng/ml and more than 5 min of a running time [11]. The bioequivalence studies for progesterone in human plasma are very limited, due to the fact of normal and variable endogenous existence levels of progesterone in human plasma, especially in the female subjects, which makes more challenge to our designed objective. To the best of our knowledge, no bioequivalence studies specifically evaluating a 400 mg intravaginal progesterone pessary formulation have been reported in the literatures, while the existing reported studies have primarily targeted different objectives, such as therapeutic drug monitoring [12], comparative bioavailability of vaginal and oral administration routes [13], the safety and impact of high-fat meal on progesterone bioequivalence [14], rectal formulation on rabbits [15], intramuscular and subcutaneous aqueous progesterone formulation [16]. Other studies have explored different objectives [17, 18]. Herein, we managed in this study to establish a new bioanalytical method to provide an accurate and reliable quantification of progesterone in human plasma, that counterbalance between a sensitive enough LLOQ with the challenge of the normal available levels of progesterone in human plasma [19, 20], taking into account to satisfy all validation requirements according to the US-FDA, EMEA and ICH guidelines, in addition to the local research center’s SOPs.
MATERIALS AND METHODS
The assay for raw material of progesterone = 99.56%, labeled progesterone as an Internal Standard (IS) (D9-progesterone, assay = 93.81%), both of which were obtained from TRC. The collected blank plasma samples were obtained for spiking from male donors at the Jordan Center for Pharmaceutical Research (JCPR) clinical site. The gradient grade (MS-quality) deionized water, acetonitrile, methanol, and ammonium formate were purchased from Fisher, Germany, while other chemicals and reagents were of analytical grade. The MS system was from Applied Biosystems MDS SCIEX API 6500+, attached to LC system from Agilent 1290 series. Analyst 1.6.3 software was installed on a PC running Windows 10 SP1 for the data management system.
The optimized HPLC conditions were fixed for mobile phase as 5 mmol ammonium formate: acetonitrile (25:75 v/v), isostatically delivered into an InertSil C8-3 (50×4.6 mm, 5 µm) column, at a stable1.0 ml/min flow rate, with the column oven temperature set to 25 °C, and the samples temperature was set al. so to 25 °C, the injection volume was fixed at 5 µland the stop of run time was set at 1.6 min. The InertSil C8-3 column was selected for progesterone analysis due to its specific retention characteristics, which provide an optimal balance between polarity and selectivity for progesterone separation from the endogenous matrix, while the most commonly used C18 columns is slightly lower in polarity, which can decrease the resolution and peak shape for progesterone. The automatically tuned MS conditions were fixed at the optimum values for progesterone detection at positive MRM scan mode: DP =105, EP =10, CE = 28, and CXP = 10. The tuned API conditions for the ion source were curtain gas = 35, CAD= gas 9, gas1 = 55, gas2 = 55, evaporation temperature = 300 °C, and the aerosol voltage = 5000 V under positive scan mode.
Master and working standard solutions
Each of progesterone and IS master standard solutions was prepared at a concentration of 1 mg/ml in absolute methanol. Then, working standard solutions were diluted from the master solution by 50% v/v methanol in water to prepare a serial dilution for plasma spiking of calibrators and Quality Control (QC) samples.
Standard calibrators and QC
Eight-level calibration points and five-level QC samples for progesterone in human plasma (pooled blank) were prepared by a single spiking step of 30 µl of working solution into 300 µl of plasma to prepare the calibrators at 3, 10, 20, 50, 90, 150, 200, and 250 ng/ml. QC levels were: LLOQ = 3 ng/ml, QC low = 9 ng/ml, QC mid1 = 30 ng/ml, QC mid2 = 100 ng/ml, and QC high = 190ng/ml. The calibrators and QC samples were prepared from two different master solutions and spiked to mimic the subjects’ matrix.
The lowest level in the dynamic range was designated as the LLOQ. The aliquots of spiked QC samples were stored in a deep freezer at −40 °C with subjects’ samples to attain the same storage conditions. During method validation and routine application, the calibration curve was constructed using a blank sample, a zero-level sample (blank spiked with IS), and eight calibration points, including the LLOQ.
Sample extraction
Progesterone with IS was extracted from human plasma samples by a protein direct precipitation technique in a single extraction step, which was used rather than liquid-liquid extraction or other multi-step extraction procedures, to minimize potential measurement variability, save time, reduce reagent consumption, and lower costs. A 300 µl of subject or standard plasma sample was quantitatively placed into an Eppendorf tube. Then 50 µl of 100 ng/ml IS and 60 µl of 0.5 M ammonium formate solution were added and vortex mixed for 30 s, then 700 µl of acetonitrile was added as a precipitation agent, and the constituents were vortexed for 30 s, then kept in a freezer under-20 °C for 15 min. After that, the mixture was vortexed again for 30 s and centrifuged for 7 min at 14000 rpm. Finally, 300 µl of supernatant was transferred to an autosampler vial for injection.
Bioanalytical method validations
The method developed for determining progesterone in human plasma was thoroughly validated in compliance with European [21] and US FDA [22] guidelines for bioanalytical method validation, with a representation of most validation sections provided here.
Specificity and carryover
The method's specificity was verified through repeated analyses of blank plasma samples from six different sources, comparing interference to the LLOQ for progesterone. The carryover effect was assessed by injecting a high-concentration sample, followed by injection of blank samples to check how efficiently the injection system can be rinsed and cleaned every injection.
Linearity, precision, and accuracy
The linear response was achieved for the peak area ratio of progesterone over IS relative to its true concentration across the established dynamic range, using a 1/x weighting factor. The slopes and intercepts from representative analytical runs of progesterone were recorded. Within-run accuracy and precision were evaluated using an analytical sequence comprising six replicates of the LLOQ and each QC sample level, with the calibration curve including both zero and blank samples. Between-run linearity, precision, and accuracy were assessed by analyzing three sets of intra-run sequences conducted on different days, with each precision run freshly prepared.
Recovery and matrix effect
The recovery of progesterone from plasma samples using the protein precipitation extraction method was assessed by comparing the peak areas of progesterone in extracted spiked QC samples (low, Mid1, Mid2, and high levels) with the corresponding peak areas in unprocessed spiking supernatant samples at the same QC levels. The Matrix effect Factor (MF) was examined in six different plasma sources for both progesterone and the IS. The MF was examined as the ratio of the peak area in the extracted blank matrix spiked with progesterone to the peak area in the corresponding quantity of progesterone in solution.
Stability
Progesterone stability was assessed in plasma and true solution for short-term and long-term of time, through a triplicate analysis for both levels of low and high QC, where each storage condition applied separately from the other storage conditions to avoid overlapping of conditions. Each stability condition was applied and analyzed in parallel with comparative freshly prepared corresponding QC samples along with freshly prepared calibrator samples. The conducted short-term stability conditions included stock solution stability at Room Temperature (R. T.) for 22 h, spiked plasma under R. T. for 13 h, three cycles of freeze-thaw, injection phase and autosampler stability at R.T for 73 h. The conducted long-term stability conditions included working solution at 2-8 °C for 29 d, spiked plasma at-20 °C for 29 d.
Clinical application and objective
The current study aimed to evaluate the bioequivalence of test product in a pilot and full study vs the Cyclogest® reference product. Where the test formulation was initially studied in six healthy female subjects, and the outcome was then confirmed in further full study of 38 subjects. Two separate clinical trials were conducted in this study to report and correlate the main PK parameters of the test product in comparison to the reference product.
Clinical design
This study was applied in accordance with the Helsinki Declaration and in compliance with good clinical practice [23]. The local Institutional Review Board (IRB) approved the study protocol before initiation under document no. IRB-01/R04. written informed consent and consent form were obtained from all participant volunteers before starting the study in a clinical site of JCPR in Amman, Jordan, in accordance with Good Clinical Practices (GCP). In each of two separate bioequivalence clinical trials, the design aimed to compare a single intravaginal dose of one pessary that containing 400 mg of progesterone under fasting conditions. This study followed an open-label, randomized, two-period, two-way crossover design to compare progesterone test formulation with the reference drug, Cyclogest®, produced by Accord-UK Ltd., United Kingdom. The final report was prepared following the ICH Topic E3 guidelines on the structure and content of clinical study reports [24], endorsed by the European Medicines Agency (EMA) [21]. A total of six healthy and adult female subjects were randomized, enrolled, and completed the pilot trial, while another 38 female subjects were enrolled and randomized in further full trial. The selection of 38 subjects for the full study was supported by regulatory guidelines and statistical power considerations specific to intravaginal progesterone formulations to ensure at least 80% power to detect bioequivalence within the specified confidence interval, considering the expected intrasubject variability in PK associated with endogenous progesterone. The selected six subjects for the pilot study aligns with common practices for preliminary assessments in bioequivalence studies [25].
Clinical procedure
Only female subjects were enrolled in this study, who met the selection criteria designed to avoid interfering from the normal and variable available progesterone levels with the administrated progesterone. The selection criteria were including healthy postmenopausal women, within age between 45-65 y, menopausal for at least one year, with an endometrial thickness of less than 5 mm. Blood hematology and blood chemistry analyses were performed during the screening and follow-up examinations. Liver function tests and Creatine Phosphokinase (CPK) assessments were conducted at the screening stage, before each study period, and during follow-up evaluations. Physical assessments and clinical evaluations were carried out at both the screening and follow-up stages. Serology and urinalysis were conducted during the screening examination. Electrocardiograms (ECG) were performed at the screening and follow-up stages. A COVID-19 PCR test was conducted on day 0 of each study period. Each study period followed a randomized crossover design included a 7 d washout phase chosen according to the terminal elimination half-life of progesterone, to avoid the carry-over effect. After a 10 h fasting period, participants received a single intravaginal dose of either the test or reference drug in pessary form. Fig. 2 summarizes briefly the flow chart for the clinical design.
PK parameters and bioequivalence statistical methods
The PK parameters of Cmax, AUC0-t, AUC0-inf, Tmax, Kel and t½ for progesterone were calculated for both test and reference products in both the pilot and full studies. The statistical analysis procedure was described in a previous study [26], where the analysis of Cmax and AUC0-t included an Analysis Of Variance (ANOVA), accounting for sequence, subject (within sequence), product, and period effects for all PK parameters, both in their untransformed state and following logarithmic transformation.90% confidence intervals and point estimates for the mean ratios of PK parameters were determined after applying logarithmic transformation to the data. The main pk parameters were considered for bio comparability design for the test product in comparison to the reference product, while AUC0-inf was assessed as a secondary parameter.
The first PK parameter taken into account was AUC0-48, which was calculated using the trapezoidal method, and the second PK parameter was the Cmax, which was derived from the plasma profile of each subject.
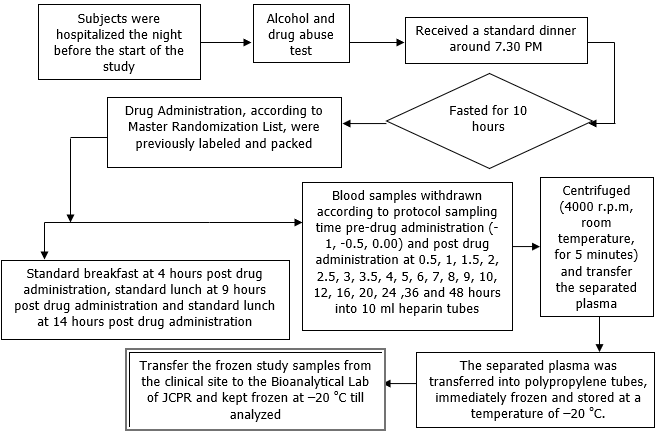
Fig. 2: The overall clinical study design flow chart
RESULTS AND DISCUSSION
LC–MS/MS analysis
The optimized tandem MS parameters exhibited high quantitative detection efficiency. The molecular ions for progesterone and IS were detected with their daughter fragment upon+MRM scan mode at the mass transition of m/z 315.197.1 and m/z 324.2113.1, respectively. The optimized chromatographic conditions were also efficient enough to separate progesterone (RT 1.29 min) from the plasma matrix with an excellent quantitative peak within a total run time of 1.6 min.
Specificity and carryover
There were interfering peaks within the acceptance criteria found at the analyte’s RT, while no interfering peaks were found at the IS’s RT, where the chromatogram for extracted plasma blank sample showed no prominent peak compared to the LLOQ. The carryover test showed that all injected blank samples subsequently to the high concentration didn’t contain any of progesterone residual peak.
Standard calibration curve and linearity
The peak area ratio of progesterone to IS in human plasma was linear over the selected dynamic range of 0.3 to 250 ng/ml. Where the mean (n=14) values for slope = 0.09099±0.00004, R2 ≥ 0.9984, and intercept = 0.00929.
Within-and between-day sensitivity (LLOQ), accuracy, and precision
The within-and between-day accuracy and precisions, including the number of replicate analysis and Standard Deviation (SD) for the analysis of progesterone in plasma (spiked QC including LLOQ) were all within the acceptance criteria, as shown in table 1.
Table 1: Summary of between days’ validation representing accuracy and precision at QC levels for progesterone
| QC | Spiked (ng/ml) |
Measured concentration | mean (ng/ml) | SD | Precision (CV%) | Accuracy (%) | |||
Within-day (n=10) |
Between-day (n=30) | Within-day (n=10) |
Between-day (n=30) | Within-day (n=10) | Between-day (n=30) | Within-day (n=10) | Between-day (n=30) | ||
| LLOQ | 0.3 | 0.25 | 0.26 | 0.03 | 0.03 | 12.18 | 9.58 | 84.05 | 88.11 |
| Low | 0.9 | 0.82 | 0.82 | 0.05 | 0.07 | 5.66 | 8.06 | 90.67 | 91.44 |
| Mid1 | 3.0 | 2.73 | 2.91 | 0.12 | 0.30 | 4.40 | 10.44 | 90.90 | 97.10 |
| Mid2 | 10.0 | 9.40 | 9.92 | 0.43 | 0.52 | 4.54 | 5.22 | 93.95 | 99.17 |
| High | 19.0 | 17.14 | 17.98 | 0.30 | 1.20 | 1.78 | 6.70 | 90.19 | 94.61 |
Recovery and matrix effect
The reported recovery values for progesterone from human plasma by acetonitrile following protein direct precipitation, as presented in table 2, demonstrated the highest extraction efficiency. This was determined by direct comparing the peak areas of extracted QC samples with those of unprocessed spiked post-extraction plasma.
Table 2: Recovery values calculated upon the mean (n=3) of peak area ratio for progesterone
| Values | (Peak Area) QC Low | (Peak Area) QC Med-1 | (Peak Area) QC Med-2 | (Peak Area) QC High | ||||
| Extracted | Post spiked | Extracted | Post spiked | Extracted | Post spiked | Extracted | Post spiked | |
| Mean n=3 | 32708 | 32055 | 99158 | 93459 | 283992 | 284048 | 590866 | 593017 |
| Recovery % | 102.04 | 106.10 | 99.98 | 99.64 | ||||
| Mean | 101.94 | |||||||
| SD | 2.969 | |||||||
| RSD% | 2.91 | |||||||
The matrix effect or MF of extracted plasma matrix on progesterone was below 12%, as well as the IS-normalized MF was less than 4%, as examined through 6 different plasma sources at both QC low and high levels and given by the mean of IS-normalized MF.
Table 3: The applied stability conditions with the corresponding results
| Validation parameters | Results for QC low (n = 3) and for QC high (n = 3) |
| Working Solution Stability at 2-8 °C for 29 d | Between 99.19 and 99.35% |
| Stock Solution Stability at R. T for 22 h | Between 97.21 and 101.76% |
| Working Solution Stability at R. T for 22 h | Between 98.21 and 100.85% |
| Short Term Stability at R. T for 13h | Between 98.47 and 102.49% |
| 3rd Cycle Freeze Thaw-Stability | Between 92.56 and 93.76% |
| Injection Phase Stability and Auto Sampler Stability at R. T for 73 h | Between 97.22 and 101.61% |
| Long Term Stability at-20 °C for 29 D | Between 94.68 and 100.01% |
Table 4: The summery for main PK parameters that calculated for both pilot and full study
| Parameter | Test | Reference | Ratio of geometric mean | 90% confidence interval (%) |
| Full study n = 38 | ||||
Cmax (ng/ml) |
Geometric mean | 15.175 | 13.359 | 113.59 |
| Arithmetic mean (n = 38) | 16.817 | 14.407 | ||
| (CV %) | 48.73 | 42.08 | ||
| SD | 8.1951 | 6.0620 | ||
AUC0-t (h*ng/ml) |
Geometric mean (n = 38) | 202.669 | 206.423 | 98.18 |
| Arithmetic mean (n = 38) | 249.875 | 234.155 | ||
| (CV %) | 70.41 | 51.95 | ||
| SD | 175.9308 | 121.6545 | ||
| Pilot study n = 6 | ||||
Cmax (ng/ml) |
Geometric mean (n = 6) | 13.094 | 11.920 | 109.36 |
| Arithmetic mean (n = 6) | 14.946 | 12.722 | ||
| (CV %) | 52.85 | 33.59 | ||
| SD | 7.8995 | 4.2731 | ||
AUC0-t (h*ng/ml) |
Geometric mean (n = 6) | 168.786 | 154.339 | 121.56 |
| Arithmetic mean (n = 6) | 211.070 | 175.274 | ||
| (CV %) | 62.42 | 43.45 | ||
| SD | 131.7537 | 76.1507 |
Stability
Progesterone stability was examined both outside and within the plasma matrix for short-and long-term conditions using QC low and high in triplicate analysis (n = 3). All results for each corresponding stability condition were above 92.5%, as shown in table 3.
Clinical application
There were no significant changes from screening regarding vital signs. ECG was performed during the screening examination, where all findings were normal for all of subjects. There were no clinically relevant abnormalities observed during the physical examination. Vital signs, blood pressure and pulse were recorded pre-dose and at scheduled time points post-drug administration. Adverse events were assessed before drug administration, at scheduled intervals post-administration, and during the washout period. The total amount of blood withdrawn during the two periods of each study was 433 ml, including samples for laboratory investigations and stock plasma. Each subject received a single intravaginal dose of one pessary, containing either the test or reference drug, under fasting conditions after a 10 h fasting period in a crossover design. In each study period, three pre-dose blood samples (3 x 8 ml) were collected at-1,-0.5, 0.00 h, followed by a series of 20 blood samples (8 ml each) collected at the times of: 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 5, 6, 7, 8, 9, 10, 12, 16, 20, 24, 36 and 48 h post drug administration. The main PK parameters for progesterone used in the bioequivalence assessment were determined from the concentration data using non-compartmental analysis. In the light of the present study, the test formulation was bioequivalent to the reference product in both the pilot and full studies, where the outcome obtained from pilot study was confirmed by the outcome obtained from the full study, demonstrating that the evaluated progesterone formulation in test product of one pessary containing 400 mg was biocomparable to the reference product of Cyclogest® 400 mg pessary. The summary of the main PK parameters for each treatment in the both bioequivalence studies is reported in table 4, calculated using non-compartmental analysis and ANOVA for progesterone.
Fig. 3 shows the concentration-time profile for the progesterone test product of 400 mg intravaginal pessary dose vs. the corresponding reference product of Cyclogest® under fasting conditions in both the pilot study (n = 6) and the full study (n = 38). The high Coefficient of Variation (CV%) for inter-subject Cmax (~48.73%) could be attributed to endogenous progesterone fluctuations, particularly in female subjects, even in postmenopausal participants. Other potential sources include differences in absorption through the vaginal mucosa and individual metabolic variations. The bioequivalent test product suggests its suitability for progesterone replacement therapy, particularly for conditions such as luteal phase deficiency, assisted reproductive technology, and hormone replacement therapy in postmenopausal women.
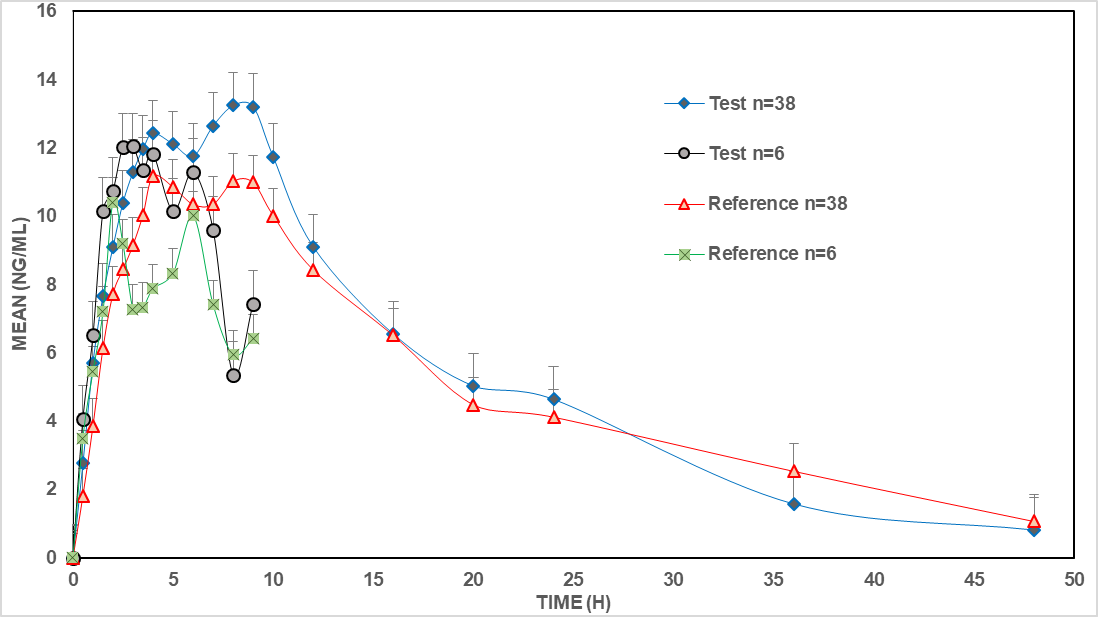
Fig. 3: The concentration-time profile for progesterone test product of 400 mg intravaginal pessary dose vs. the corresponding reference product of Cyclogest®under fasting conditions in both the pilot study (n = 6) and the full study (n = 38)
The current results are in agreement with the previous research in the field, where a study by [27] compared the efficacy, safety, and tolerability of progesterone vaginal pessaries (400 mg twice daily) to progesterone vaginal gel (90 mg once daily) for luteal phase support in women undergoing IVF. The clinical pregnancy rates were comparable between the two groups, indicating similar efficacy. Additionally, both treatments were found to be safe and well-tolerated by patients. As a result, our study provides robust evidence for the bioequivalence of a newly developed 400 mg intravaginal progesterone pessary compared to Cyclogest®, and contributes valuable PK data for regulatory evaluation and clinical application, particularly in postmenopausal women. Additionally, future studies could utilize the current results as a reference for further PK applications under different clinical conditions, such as co-administration with other hormones or in patients with hepatic or renal impairment, and explore the application of the test intravaginal progesterone formulation in diverse populations, such as premenopausal women or pregnant individuals requiring luteal phase support.
CONCLUSION
A selective and sensitive enough bioanalytical methods for the quantitative determination of Progesterone was developed and validated, then successfully applied for critical evaluation of PK and bioequivalence studies. The validated method included the recommended procedures to demonstrate that the analytical method for the plasma matrix is reliable and reproducible according to US-FDA, EMEA and ICH guidelines. In pilot study, post administration of one pessary that contains 400 mg intravaginal progesterone for both test and reference products, the test product has showed a bioequivalent PK parameters to the reference product. Then, the test product was evaluated again in full study to confirm the obtained results from the pilot study, where the overall outcome for the test product is perfectly bioequivalent to the reference product of Cyclogest®. This study helps fill a gap in current research by providing strong evidence that the 400 mg intravaginal progesterone pessary is bioequivalent to the reference product. The validated LC-MS/MS method allows for accurate progesterone measurement, even with natural hormonal fluctuations. Clinically, these findings suggest that this formulation could be a reliable option for women who need progesterone supplementation for reproductive health and hormone therapy.
ACKNOWLEDGMENT
Many thanks all for the working team in the JCPR.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
Tawfiq Arafate: Investigation, project administration, formal analysis, resources, principal supervision. Ahmed Abu-awwad: writing original draft, methodology, validation. Basil Arafat: data curation, visualization, Conceptualization, software, review and editing.
CONFLICTS OF INTERESTS
The authors declare no conflict of interest
REFERENCES
Kumar P, Magon N. Hormones in pregnancy. Niger Med J. 2012;53(4):179-83. doi: 10.4103/0300-1652.107549, PMID 23661874.
Lobo RA. The role of progestins in hormone replacement therapy. Am J Obstet Gynecol. 1992;166(6):1997-2004. doi: 10.1016/0002-9378(92)91401-U, PMID 1605291.
Unfer V, Casini ML, Marelli G, Costabile L, Gerli S, Di Renzo GC. Different routes of progesterone administration and polycystic ovary syndrome: a review of the literature. Gynecol Endocrinol. 2005;21(2):119-27. doi: 10.1080/09513590500170049, PMID 16109599.
Fatemi HM, Popovic Todorovic B, Papanikolaou E, Donoso P, Devroey P. An update of luteal phase support in stimulated IVF cycles. Hum Reprod Update. 2007;13(6):581-90. doi: 10.1093/humupd/dmm021, PMID 17626114.
Levine H, Watson N. Comparison of the pharmacokinetics of crinone 8% administered vaginally versus prometrium administered orally in postmenopausal women. Fertil Steril. 2000;73(3):516-21. doi: 10.1016/s0015-0282(99)00553-1, PMID 10689005.
Paulson RJ, Collins MG, Yankov VI. Progesterone pharmacokinetics and pharmacodynamics with 3 dosages and 2 regimens of an effervescent micronized progesterone vaginal insert. J Clin Endocrinol Metab. 2014;99(11):4241-9. doi: 10.1210/jc.2013-3937, PMID 24606090.
Cometti B. Pharmaceutical and clinical development of a novel progesterone formulation. Acta Obstet Gynecol Scand. 2015;94 Suppl 161:28-37. doi: 10.1111/aogs.12765, PMID 26342177.
Miles RA, Paulson RJ, Lobo RA, Press MF, Dahmoush L, Sauer MV. Pharmacokinetics and endometrial tissue levels of progesterone after administration by intramuscular and vaginal routes: a comparative study. Fertil Steril. 1994;62(3):485-90. doi: 10.1016/S0015-0282(16)56935-0, PMID 8062942.
Cicinelli E, Cignarelli M, Sabatelli S, Romano F, Schonauer LM, Padovano R. Plasma concentrations of progesterone are higher in the uterine artery than in the radial artery after vaginal administration of micronized progesterone in an oil based solution to postmenopausal women. Fertil Steril. 1998;69(3 Pt 2):471-3. doi: 10.1016/s0015-0282(97)00545-1, PMID 9531879.
Albayrak M, Kadioglu Y, Demirkaya Miloglu F, Borekci B. A simple HPLC method for the determination of plasma progesterone levels in the third trimester of human pregnancy. Lab Med. 2024;19:lmae098. doi: 10.1093/labmed/lmae098, PMID 39703165.
Tran CS, Bui QD, Nguyen NT, Dao MH, Nguyen TT. LC-MS/MS method for rapid quantification of progesterone in rabbit plasma and its application in a pharmacokinetic study of the transdermal formulation. J Anal Methods Chem. 2020;2020:8889375. doi: 10.1155/2020/8889375, PMID 33178479.
Mihu D, Vlase L, Imre S, Mihu C, Achim M, Muntean D. New LC/MS method for determination of progesterone in human plasma for therapeutic drug monitoring in pregnancy and gynecological disorders. Stud Univ Babes Bolyai Chem. 2009;54(1):151-60.
Wang H, Liu M, Chen R, Deng C. Clinical re-evaluation on bioequivalence and relative bioavailability of micronized progesterone hard capsule (Yimaxin) and micronized progesterone soft capsule (Utrogestan) under vaginal and oral administration routes. Pak J Med Sci. 2021;37(7):1740-6. doi: 10.12669/pjms.37.7.3949, PMID 34912388.
Qin H, Zheng L, Fang Y, Liu Y, Liu A, WU J. Pharmacokinetics safety and bioequivalence of two formulations of progesterone soft capsule in healthy Chinese postmenopausal females: impacts of a high-fat meal. Basic Clin Pharmacol Toxicol. 2022;130(2):268-76. doi: 10.1111/bcpt.13687, PMID 34806331.
Long L, Huang Q, WU M, Hou S, Dai Z. Bioequivalence of progesterone sustained release suppository in rabbits. J Huazhong Univ Sci Technolog Med Sci. 2005;25(4):470-2. doi: 10.1007/BF02828227, PMID 16196307.
Mehta S, Chaturvedi A. Comparison of the pharmacokinetics bioequivalence and safety of aqueous progesterone formulation administered as either intramuscular or subcutaneous injection versus oil-based progesterone formulation administered as intramuscular injection: a randomized study. J Clin Diagn Res. 2023;17(1):1-6.
Patil N, Maheshwari R, Wairkar S. Advances in progesterone delivery systems: still work in progress? Int J Pharm. 2023 Aug 25;643:123250. doi: 10.1016/j.ijpharm.2023.123250, PMID 37481096.
Alambiaga Caravaca AM, Gonzalez Iglesias LG, Rodilla V, Kalia YN, Lopez Castellano A. Biodistribution of progesterone in the eye after topical ocular administration via drops or inserts. Int J Pharm. 2023;630:122453. doi: 10.1016/j.ijpharm.2022.122453, PMID 36455753.
Fassorra C, Luisi M. The measurement of progesterone in human plasma by competitive protein binding radioassay. Hormones. 1971;2(3):153-63. doi: 10.1159/000178231, PMID 5006658.
European Agency for the Evaluation of Medicinal Products. Progesterone: summary report. Eval Inspect. 1999;1-4.eu.int.
Committee for Human Medicinal Products. EMA/CHMP/ICH. ICH guideline M10 on bioanalytical method validation; 2019.
Food and Drug Administration. Bioanalytical method validation guidance. In: FDA. 2018 May;25. Available from: https://www.fda.gov/files/drugs/published/bioanalytical-method-validationguidance-for-industry.pdf.
World Medical Association (WMA). Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. Jahrbuch fur Wissenschaft und Ethik. 2009;14(1):233-8. doi: 10.1515/9783110208856.233.
International council for harmonisation of technical requirements for pharmaceuticals for human use. E3: structure and content of clinical study reports. ICH; 1995. p. 49.
FDA. Guidance. Bioequivalence studies with pharmacokinetic endpoints for drugs submitted under an ANDA. FDA Guid. 2021 Aug;24.
Omari KW, Abu Awwad A, Arafat B, Macovei G, Boltez TL, Abdel Baki ZA. Simultaneous quantification of ramipril and ramiprilat in drug formulations: A clinical LC-MS/MS study. Indonesian J Pharm. 2024;35(3):512-20. doi: 10.22146/ijp.9302.
Saunders H, Khan C, D Hooghe T, Magnusdottir TB, Klingmann I, Hrafnsdottir S. Efficacy safety and tolerability of progesterone vaginal pessaries versus progesterone vaginal gel for luteal phase support after in vitro fertilisation: a randomised controlled trial. Hum Reprod. 2020;35(2):355-63. doi: 10.1093/humrep/dez261, PMID 32074281.