
Int J App Pharm, Vol 17, Issue 4, 2025, 521-528Original Article
FORMULATION AND EVALUATION OF NANO FORMULATION OF BTK INHIBITOR BY BOX-BEHNKEN DESIGN AND HIGH-PRESSURE HOMOGENIZATION FOR ENHANCED BIOAVAILABILITY AND REDUCING THE EFFECTS OF FOOD
S. SREENIVASA CHARY1, D. V. R. N. BHIKSHAPATHI1,2*, V. V. RAJESHAM3, SAILAJA RAO PENAKALAPATI2, PAMU SANDHYA4, RUBESH KUMAR SADASIVAM2
1Bir Tikandrajit University, Canchipur, Imphal West-795003, Manipur, India. 2Teegala Ram Reddy College of Pharmacy, Meerpet, Hyderabad–500097, India. 3Department of Pharmacology, CMR College of Pharmacy, Kandlakoya (V) Medchal (MandD), Hyderabad-501401, Telangana, India. 4Shadan Women’s College of Pharmacy, Khairatabad, Hyderabad-500004, Telangana, India
*Corresponding author: D. V. R. N. Bhikshapathi; *Email: dbpathi71@gmail.com
Received: 28 Feb 2025, Revised and Accepted: 05 May 2025
ABSTRACT
Objective: The study aims to enhance the solubility and oral bioavailability of the poorly soluble drug Ibrutinib (IBR), a type IV irreversible kinase inhibitor, using Nanosuspension (NS) as a formulation strategy. The study also evaluates the ability of NS to minimize fasted-fed variability and improve drug absorption.
Methods: NS was prepared using High-Pressure Homogenization (HPH) and optimized with a Box-Behnken design. Poloxamer 188 and Hydroxypropyl Methylcellulose (HPMC E-15) were used as stabilizers to prevent particle aggregation. A comprehensive evaluation of the formulation was conducted, including particle size analysis, zeta potential measurement, Scanning Electron Microscopy (SEM) for shape determination, drug-excipient interactions, in vitro dissolution studies, and in vivo pharmacokinetic assessments in both fed and fasting conditions.
Results: The particle size of the optimized NS ranged from 134.6 to 214.0 nm, with polydispersity indices between 0.189 and 0.56, indicating a uniform size distribution. SEM confirmed the nanosized particles with a stable morphology. IBR-NS exhibited an 18.45-fold increase in solubility compared to pure IBR, reducing precipitation and enhancing intestinal absorption. Pharmacokinetic studies demonstrated a 3.05-fold increase in concentration max and a 3.97-fold increase in area under the curve0–t under both fed and fasting conditions. Notable differences in pharmacokinetics between the fed and fasted states were observed, supporting improved drug absorption.
Conclusion: The study confirms that NS is a promising approach for improving IBR solubility and bioavailability. The NS formulation effectively reduces fasted-fed variability, enhances intestinal absorption, and holds potential for better therapeutic outcomes.
Keywords: Ibrutinib, Nanosuspension, HPMC E15, Poloxamer 188, Bioavailability
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i4.54079 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
The cure of MCL (Mantle Cell Lymphoma) and CLL (Chronic Lymphocytic Leukaemia) has been revolutionized by the irreversible Bruton's Tyrosine Kinase (BTK) inhibitor Ibrutinib (IBR). With a strong IC50 of 0.5 nM, IBR blocks the B-Cell Receptor (BCR) pathway [1]. IBR binds to BTK and has varying affinity for kinases, including BLK, JAK3, EGFR family, and TFK and effective in treating malignancies [2]. IBR, although showing promise in B-cell malignancies, reported with poor solubility and low oral bioavailability of 2.9%. This restriction is linked to its first-pass metabolism susceptibility and low solubility (0.002 mg/ml) [3]. The daily dosage is higher for treating leukaemia, leading to serious adverse effects on the nervous system. In addition to these problems, when food is given with IBR, there is an almost two-fold spike in exposure [4] affecting drug's safety and efficacy. Therefore, a strategy that reduces the pharmacokinetic variations between the fed and fasting states must be developed. Strategies such as phospholipid complexes, Nanosuspension (NS), Nanostructured Lipid Carriers (NLCs), and Self-Nanoemulsifying Drug Delivery Systems (SNEDDS) are designed to improve the bioavailability of IBR while addressing fast-fed variability. Scalability problems and the need for pricey specialist excipients are obstacles [3, 5–7]. IBR displayed pH-dependent solubility and precipitation is brought on by the passage of food from the stomach to the intestinal areas. So, a NS formulation is more suitable to enhance the solubility of this high-dose medication at gut pH. Breakthroughs in NS face challenges related to scalability when using lab-scale methods. We aim to get beyond this restriction by applying commercially feasible techniques, particularly High-Pressure Homogenization (HPH).
NS have shown promise in improving bioavailability and reducing fast-fed variability for anti-cancer drugs [8, 9]. NS stabilized by additives, are created through top-down, bottom-up, or hybrid methods. Bottom-up methods are economical, easy to use, and do not require specialized tools. However, they have trouble managing the particles' size, which causes instability and non-uniformity problems [10].
The top-down technique employs high-energy actions for Particle Size (PS) reduction, such as milling or HPH. Despite their industrial utility, these techniques incur high costs and extended processing times. However, optimization considering both formulation and process variables can enhance time efficiency. This study aimed to formulate NS to improve oral bioavailability and decrease fast-fed variability using poloxamer 188 and HPMC E15 as stabilizers, a HPH to a 3-level and 3-factor Box Behnken design (BBD) was applied. The characterization process involved Fourier Transform Infrared Spectroscopy (FTIR), Differential Scanning Calorimetry (DSC), and Scanning Electron Microscopy (SEM) investigations. Dissolution test and pharmacokinetic assessment under fasting and fed settings were performed.
MATERIALS AND METHODS
Materials
Dr Reddy’s Laboratories Pvt. Ltd., Hyderabad, India, provided us with sorafenib and IBR. Sigma-Aldrich® in India provides a variety of chemicals, such as Pluronic F-127, poloxamer 188, TPGS (Tocopheryl Polyethylene Glycol Succinate), Sodium taurocholate, and pepsin. Other materials included mannitol, Tween®80, HPMC E-15 (hydroxy propyl methylcellulose), acetone, acetonitrile, trehalose, and sucrose that were acquired from TCI Chemicals, and Alpha Aesar, India.
Methods
Analytical and bioanalytical method development using reverse-phased high-performance liquid chromatography
IBR was analyzed chromatographically using a Reverse-Phase High-Performance Liquid Chromatography (RP-HPLC) method on a Develosil ODS HG-5 RP C18 (150 x 4.6 mm, 5µm). The mobile phase is composed of 0.1% orthophosphoric Acid and methanol in the ratio of 35:65. The flow rate employed was 1.0 ml/min. At 287 nm, detection was carried out. The technique showed linearity within the scale of 25-1000 ng/ml [7]. Protein precipitation was used to remove IBR from plasma trials. In summary, 250 µl** of acetonitrile was used to quench 50 µl** of rat plasma containing sorafenib as the internal standard. The mixture sample was vortexed and centrifuged for 10 min at 10,000 rpm. RP-HPLC was used to separate and examine the supernatant at 287 nm.
Formulation and optimization of NS using HPH
IBR NS was formulated by dispersing the drug in stabilizers (poloxamer 188 and HPMC E15) followed by subjected to HPH (Microfluidizer, HPH; LM 20). In brief the procedure is as follows: Weighted stabilizers were thawed in double distilled water (100 ml), and IBR was mixed under stirring at an rpm of 500 for 30 min using high shear homogenization of Ultra Turrax model T25, Make: IKA). The coarse suspension was subjected to a pressure homogenizer for 20 min at 1000 bar in continuous means under ice-cold conditions. The NS was lyophilized at-54 ℃ under vacuum (FD-01, Delvac Pumps Pvt. Ltd.) using mannitol (5 % w/v) as a cryoprotectant and stored for further findings [11].
Optimization supported by design-expert software
The stabilizer concentration significantly impacts the creation of a stable NS. In addition, homogenization pressure and the number of cycles were found to affect PS reduction [9]. To thoroughly investigate the effects of formulation and process factors on the responses of NS, a three-factor, three-level Box-Behnken Design (BBD) was employed [12]. The study factors along with responses (PS and Poly-Dispersity Index; PdI), as detailed in table 1. An investigational design was built using Design-Expert TM package (version 13.0.0.1, Stat-Ease Inc., Minneapolis, MN).
Table 1: Runs designed for the trails (drug in each run is 40 mg)
| Run | Factor 1 | Factor 2 | Factor 3 | Response 1 | Response 2 |
| A: Stabilizer conc. (w/w) | B: Cycles (no) | C: Pressure (bar.) | Particle size | PdI | |
| ratio | rpm | min | nm | ||
| 1 | 20 | 1 | 1000 | 185 | 0.426 |
| 2 | 40 | 3 | 1000 | 178 | 0.32 |
| 3 | 20 | 3 | 1500 | 150 | 0.29 |
| 4 | 20 | 5 | 1000 | 172 | 0.41 |
| 5 | 60 | 5 | 1000 | 140 | 0.28 |
| 6 | 40 | 3 | 1000 | 201 | 0.42 |
| 7 | 40 | 3 | 1000 | 200 | 0.44 |
| 8 | 60 | 3 | 500 | 153.8 | 0.33 |
| 9 | 20 | 3 | 500 | 203 | 0.56 |
| 10 | 40 | 1 | 1500 | 158 | 0.33 |
| 11 | 40 | 5 | 500 | 202 | 0.56 |
| 12 | 40 | 5 | 1500 | 151 | 0.189 |
| 13 | 60 | 1 | 1000 | 149 | 0.28 |
| 14 | 40 | 1 | 500 | 214 | 0.45 |
| 15 | 40 | 3 | 1000 | 199 | 0.38 |
| 16 | 40 | 3 | 1000 | 200 | 0.49 |
| 17 | 60 | 3 | 1500 | 134.6 | 0.198 |
Using a One-Factor-At-A-Time (OFAT) model, the numeral of homogenization cycles vital to achieve a mean size in the range of 100–500 nm at 1500 bar pressure was optimized. First, a pre-homogenized suspension was made using the procedure outlined. After applying one to five homogenization cycles at 1500 bar, distribution of size and PS studies were utilized to modify the formulation [13].
Evaluation of NS
Measurements of size, polydispersity index and potential
Size of the formed IBR NS, and potential were assessed by Dynamic Light Scattering (DLS) concept with a zeta sizer (nano ZS, United Kingdom). The trials were suitably diluted with deionized water (10 times) before investigation at 25 °C [14].
FTIR
FTIR (Perkin Elmer) was used to look for possible drug-stabilizer interactions. Drug, the physical combination, and the freeze-dried NSs were all examined [15].
DSC
DSC was used to analyse thermal behaviour (Mettler Toledo DSC-1). Samples weighing one and two milligrams were put in aluminium pans and heated at 10 degrees celsius per minute from 25 to 200 °C [16].
Topography
The outer structure of the drug and NS was snapped using FESEM (Quanta 250 SEM). The drug and NS were given thin coating of Au with an ion sputter, and then positioned over double-sided carbon adhesive tape. Mounted the tape over aluminium pin counterfoils, before the actual examination. The samples were tested at a working distance of 10 mm, 500–10,000 times magnification, and 30 kV accelerating voltage [17].
Solubility Investigations
The drug, and the physical mixing of the drug with stabilizer and freeze-dried NS equivalent to or freeze-dried NS, physical mixture (poloxamer 188 and HPMC E15) and PD equivalent to 4 mg drug were separately transferred to the screw vials (4 millilitre) containing deionized water. The bottles were then subjected to sonication for three minutes, whose aim was getting uniform dispersion of the drug. All the vials were then placed on the SI 200D incubation shaker located in the UK for 72 h at 37 °C. Final centrifugation (Thermo-Fisher Model) at 8000 rpm for 10 min was done. The resulting clear solution was separated and filtered using a Millipore 0.22 filter, and the drug content was quantified using the established analytical method [18].
Dissolution studies
Drug release studies were carried out in Fed State Simulated Gastric Fluid (FeSSGF), Fed State Simulated Gastric Fluid (FeSSIF), Fasted State Simulated Gastric Fluid (FaSSGF), and Fed State Simulated Intestinal Fluid (FeSSIF) [18]. IBR equivalent to 500 µg was utilized for other mediums, while 50 mg was added for FaSSGF (because of its solubility of 2 mg/ml). In vitro release experiments of the NS and pure drug were carried out in a paddle type of USP model DS 1400 (Lab-India) at 50 rpm in 250 ml of dissolution media at a 37 °C±0.5 °C. Three-millilitre samples (3 ml) were taken at prearranged intervals to maintain sink conditions, and an equivalent part of fresh media was supplied. Sigma-Aldrich 0.1-µm nylon membrane syringe filters were used to filter the collected samples, and analysed the samples by UV visible Spectrophotometer. A comparison was made between pure drug and NS. There were three study runs carried out [19].
Stability studies
The optimized and FD-NS (FD-NS) were stored for 3 mo at three different temperatures as per ICH guidelines by storing at high temperature (40 °C), room temperature (28±2 °C), and at refrigerator conditions (4–8 °C). The optimised preparation was tested for PS, PdI, and ZP on the 0th, 15th, one month, and three months using a standard zeta-sizer.
Pharmacokinetics
Animal studies
Wistar rats, which for the study were purchased from the Nutrition National Institute (NIN), Hyderabad, India. The weighing of rat was approximately 220±20 g and at an age of around 4 to 5 w. This investigation has been accepted by (IAEC), under no.1447/PO/RE/S/11/CCSEA-80/A. Before the experiment, rats were exposed to acclimatization of controlled environment that simulated the natural light-dark cycle, (20 °C±2 °C and a RH of 40–60%) for a one-week period.
Rats were allocated into 4 sets randomly, with each group comprising six animals. Oral administration of the NS formulation at 40 mg/kg body weight, the vehicle, or IBR dissolved in 0.25% w/v HPMC. Blood samples of 250 µl** withdrawn from the retroorbital plexus at defined time points and transferred to EDTA-coated tubes. The samples were centrifuged in Eppendorf centrifuge at 6500 rpm for 12 min; then the plasma was assessed by HPLC as per the developed method. Data analysis was done using the non-compartmental method determining pharmacokinetic parameters using Win Nonlin version 3.1; Pharsight Co., Mountain View, CA, USA.
RESULTS AND DISCUSSION
Analytical and bioanalytical method development using reverse-phased high-performance liquid chromatography
RP-HPLC carried out by performing analysis at 287 nm, and R2 was 0.997. Specificity was established through chromatogram comparison for the sample and a blank. Response in the biological medium was related with that of a pure standard to determine the percentage retrieval. For removal of IBR from plasma trials, a precipitation of protein technique using 250 µl** of solvent (acetonitrile) was adopted.
Formulation and optimization of NS using HPH
When it comes to low solubility drugs, the PS, PdI, and ZP were considered as crucial physical properties. Increasing the Surface Area (SA) and decreasing PS using HPH, which uses a high energy input, helps improve active substances' solubility and bioavailability. The present study formulated NS using HPH microfluidics. Several surfactant stabilizers like poloxamer 188, pluronicsF-127 (PF 127), tween 80 (polysorbate 80) and Tocopheryl Polyethylene Glycol Succinate (TPGS) and a polymeric stabilizer like HPMC E15 were tested to see if they could produce a stable NS. The effects of each stabilizer on PS and PdI were evaluated while testing them at constant ratio (0.2% w/v) and constant process variables (number of cycles three and pressure of 500 bar). In the succeeding direction: poloxamer 188>PF 127>polysorbate 80>TPGS>HPMC E-15, reduced the size of NS. When compared to surfactants with medium HLB values (Tween 80, TPGS), the PS of IBR-NS was larger when using surfactants with low and high HLB values (PF-127 and TPGS). In addition, the polymer HPMC E15 reduced the PS of IBR-NS, and the formulations' zeta potentials went as follows: poloxamer 188>tween 80>pluronics F 127>HPMC E15>TPGS.
These findings clearly showed that stabilizer type affects both ZP and PS. But, as demonstrated by increased PS and high PdI, the suspension made with a single stabilizer was not actually stable (with the exception of Poloxamer 188) for longer than four days, which may be related to the Ostwald ripening process [20]. In order to create a steady formulation with a decreased PdI and PS, a set of stabilizers was used. Poloxamer 188 and HPMC E15 were used as combination to inhibit crystal growth and to stabilize the system through intermolecular interfaces, specifically hydrogen bonding between the IBR and HPMC E-15. While poloxamer 188 prevents agglomeration by creating a shield around the nanoparticles and applying repulsive and steric hindrance. Doses of the stabilizer (1:1 ratio) ranged from 20% to 60%. In the current study, seventeen trails formulation with five centre points were taken up and subjected to multiple linear regression analysis models. The model assortment process uses regression coefficient and coefficient of variance with Analysis of Variance (ANOVA) to evaluate the impact of variables.
PS
The PS ranged from 134.6 to 214 nm, with a PdI that varied from 0.189 to 0.56. Fig. 1 displays surface response and contour plots that represent the effects of study factors on the PS. The F-value of 15.14 indicates that the chance of interference from noise is less than 0.08%, validating the relevance of the model as quadratic with minimal LOF (Lack of Fit). From ANOVA, variables whose p-value is less than 0.0500 result in the model's R², adjusted R², and predicted R² of 0.9511, 0.8883, and 0.7236, correspondingly. The two main terms with significant terms-A, C, and A² were responsible for getting obtained regression equation.
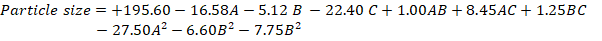
PdI
The PdI stands for the dimensionless measure of width for the size distribution of the particles, usually ranging between 0 and 1. Formulations prepared yielded PdI in the range of 0.189-0.561. An F-value of 12.29 indicates a mere 0.04% probability that the model's significance arises from random variation, thus confirming its status as a significant linear model with minimal lack of fit. A value of F 0.94 indicates that the lack of fit is not statistically noteworthy.
In order to construct the model, certain non-significant variables were eliminated after an ANOVA revealed significant components with a p-value<0.0500. Regression Coefficients: R², adjusted R², and predicted R² valued at 0.7394, 0.6792, and 0.5678 correspondingly depict satisfactory precision of 12.269, which goes beyond the required amount that must be achieved as 4 with variables A and C showing the most impact.

Positive coefficients indicated a direct relationship, and an increase in the relevant variable(s) increases the PdI. In contrast, the negative coefficient of-0.0155 implies that a decrease in the associated variable(s) reduces the PdI. The response surface plots in fig. 1 show that the velocity of homogenization impacts the PdI to a great extent.
After analysing the results of the runs given by design, an optimized solution was generated with a desirability values. The formulation selected (F opt solution) had an extreme attractiveness nearer to 1.0. The optimum constraints were the stabilizer of 59.99 (24 mg of stabilizer for 40 mg of the medication), the cycles of 2.44, and 1499 as pressure. Fig. 2 represented the design space along with overlay plot. The projected PS and PdI were 130.235 nm, 0.189 respectively; the corresponding observed mean values were at 130±13.3 nm and 0.182±0.013, respectively.
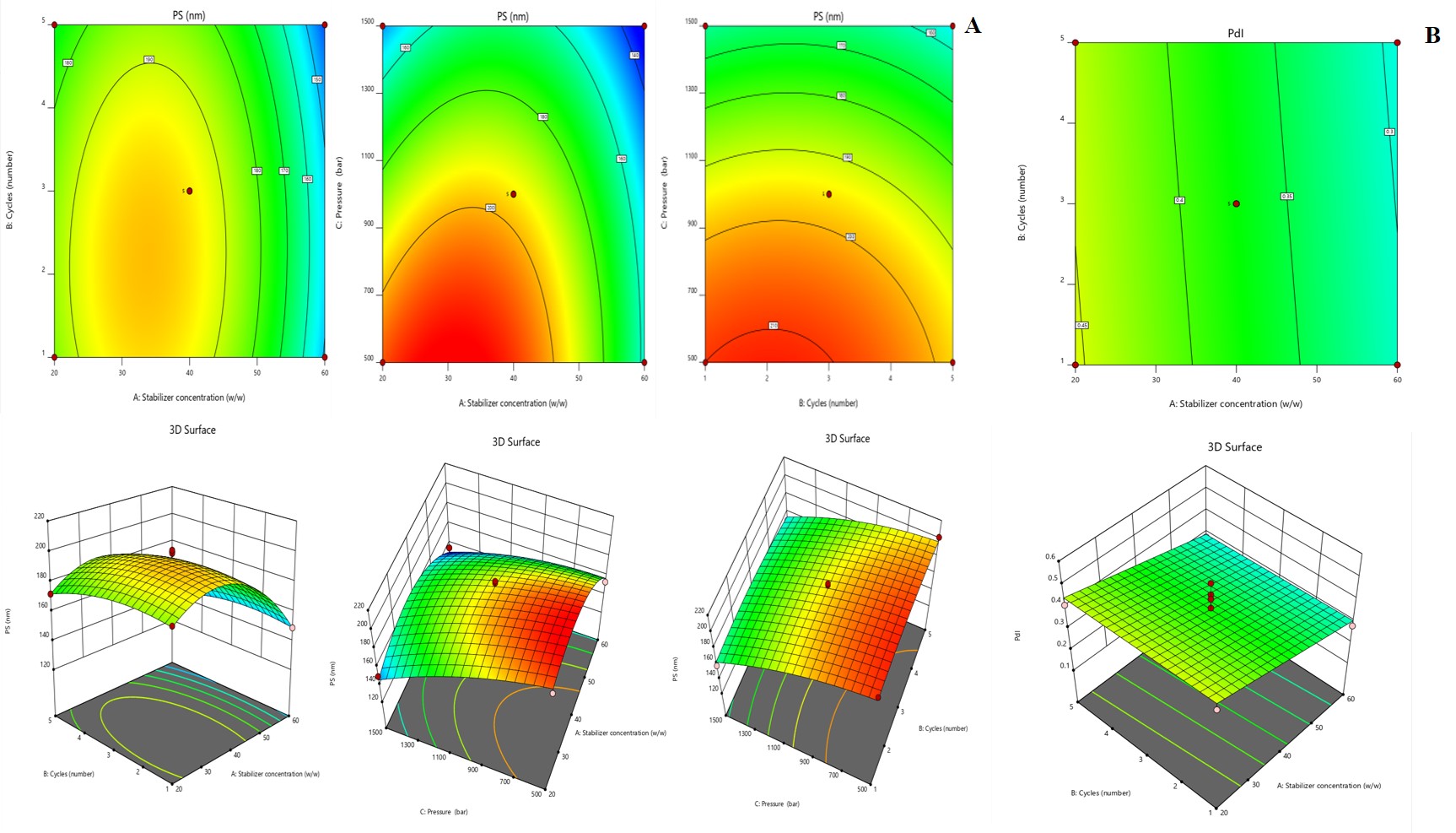
Fig. 1: A) Graphical representation of response surface and contour plots showing the effect of variables on particle size, B) SR and contour plots (CP) demonstrating the consequence of variables on PdI
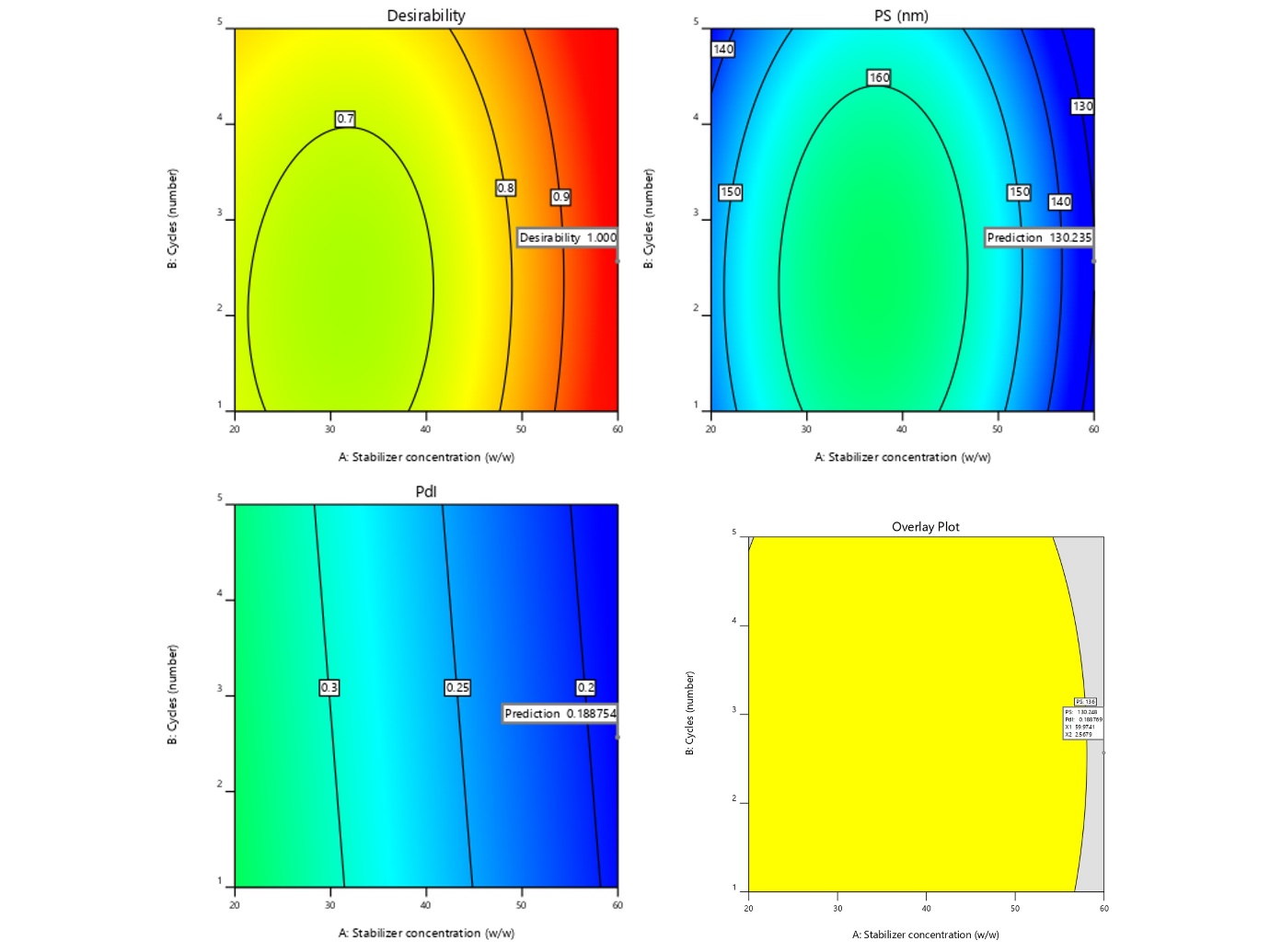
Fig. 2: Graphical representation of desirability and Overlay plot (yellow area denotes the feasible region)
Evaluation
PS, PdI, and ZP
The size and PdI of the prepared NS were 130±13.3 nm, 0.182±0.013, indicating the homogeneousness of the system. Regarding stability, the zeta potential of the particles is a very important measure because it is related to the Double Electric Layer (DEL) surrounding them. It had a zeta potential of 13±5.6 mV that was down due to a polymeric coating of the stabilizer. From this, it was concluded that rather than sterically interacting ones, electrostatically stabilized high-zeta-potential nanoparticles are chiefly required (fig. 3).
Topography
Fig. 3 showed the morphology of drug and NS surface characteristics. The drug has a broad PS with discrete irregular cubic form in µm size. Nevertheless, the NS formulation exhibited spherical nanosized particles with a size of less than 200 nm, and the results correlate with the zeta sizer.
Fig. 3: Malvern image of A) Particle size and PdI; B) ZP; C) SEM image of pure drug; D) Nanosuspension (NS)
FTIR
Analyzing the nano formulation, excipients, and simple IBR IR spectra was used to determine component compatibility, as shown in fig. 4. 400–4000 cm–1 was the scanning range used. The simple drug showed distinct peaks at (1241, 1147, 1100, 986, and 953 cm-1). As no new peaks were seen in the NS-FD preparation and PM it clearly indicates no chemical interaction between the stabilizer and medication and they are compatible [21].
DSC
The following fig. 4 reveals the DSC analysis of IBR, the stabilizer's physical combination, and the NS: The crystalline nature of drug was confirmed by a strong melting peak at 158 °C, whereas the stabilizer demonstrated a peak at 58.96 °C. These findings align with prior studies, indicating a crystalline-to-amorphous transition aided by precipitation and the drug is trapped inside or coated by the stabilizer [6].
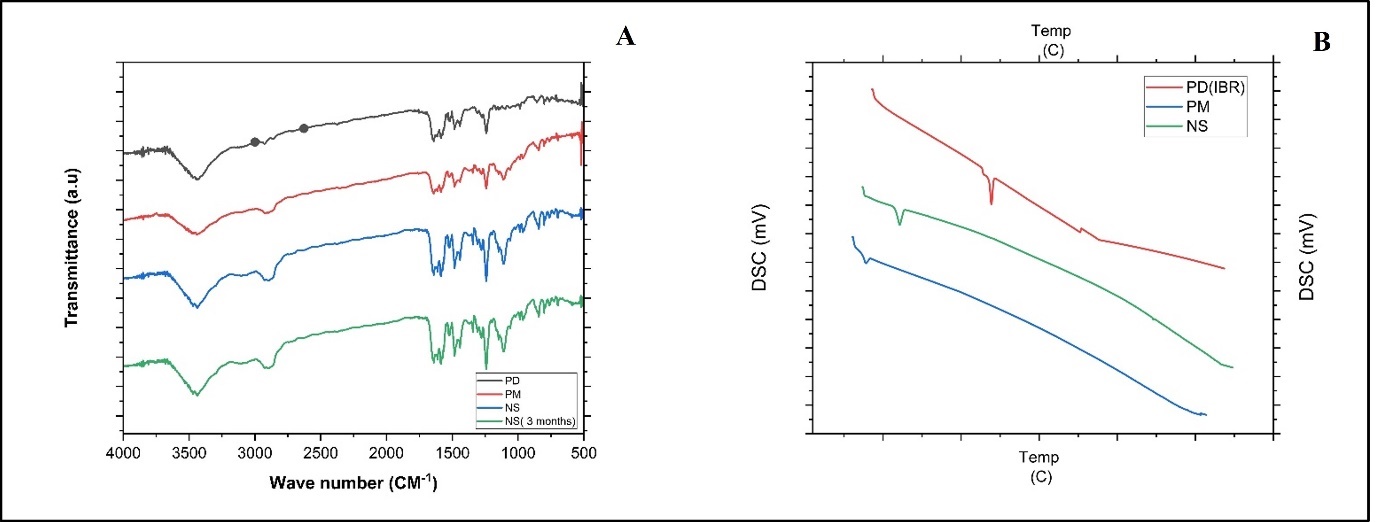
Fig. 4: A) Overlay of FTIR analysis PD (black line-IBR; PM (red line-Physical mixture; NS (Blue line-Nanosuspension (NS); and D) NS kept at room temperature for 3 mo (Green line). B) Overlay of DSC thermograms of PD (red line IBR); PM (Blue line-Physical mixture, NS (Green line-Nanosuspension)
Solubility investigations
Solubility investigations were carried out. In case of NS drug solubility was 78.24± 6.68 µg/ml, 8.28 ±1.186 µg/ml in case of physical mixture, and the plain drug solubility was 4.22±0.055 µg/ml. NS is showing an 18.45-fold high solubility for drug in comparison to PD. The reason behind is ascribed to the increased wetting of the drug, decreased size and as well no agglomeration promoting drug dispersion and dissolution. It clearly suggested that the stabilizers' alone presence cannot achieve the solubility enhancement as observed in the NS formulation. Instead, it is the combination of reduced PS and the presence of stabilizers that synergistically contribute to the observed improvement in solubility [22].
Dissolution studies
The dissolution profile of the NS and PD in both FaSSGF, FeSSGF, FaSSIF, and FeSSIF media, is shown in fig. 5. In the fed state gastric fluid, the PD disclosed only 19.66±2.4% release in 2 h, however the NS displayed 86.72±2.66% drug release. In fasted state gastric and intestinal media, PD release 69.92 ±4.15% and 57.33±2.98%, while the formulation dissolved 97.04±4.66 % and 98.62±1.66 % after 2h. Both formulations displayed comparable drug release characteristics in FaSSGF, likely due to IBR's strong solubility in acidic pH. The enhanced drug release from the pure drug in FeSSIF may be explained by the different compositions of media in fast and fed state. Instead, the drug release from the formulation was constant in both medias. The Noyes-Whitney/Nernst-Brunner equation favours higher dissolution due to a reduction in particle to the nano range, which is responsible for the enhanced drug release from the NS [23, 24].
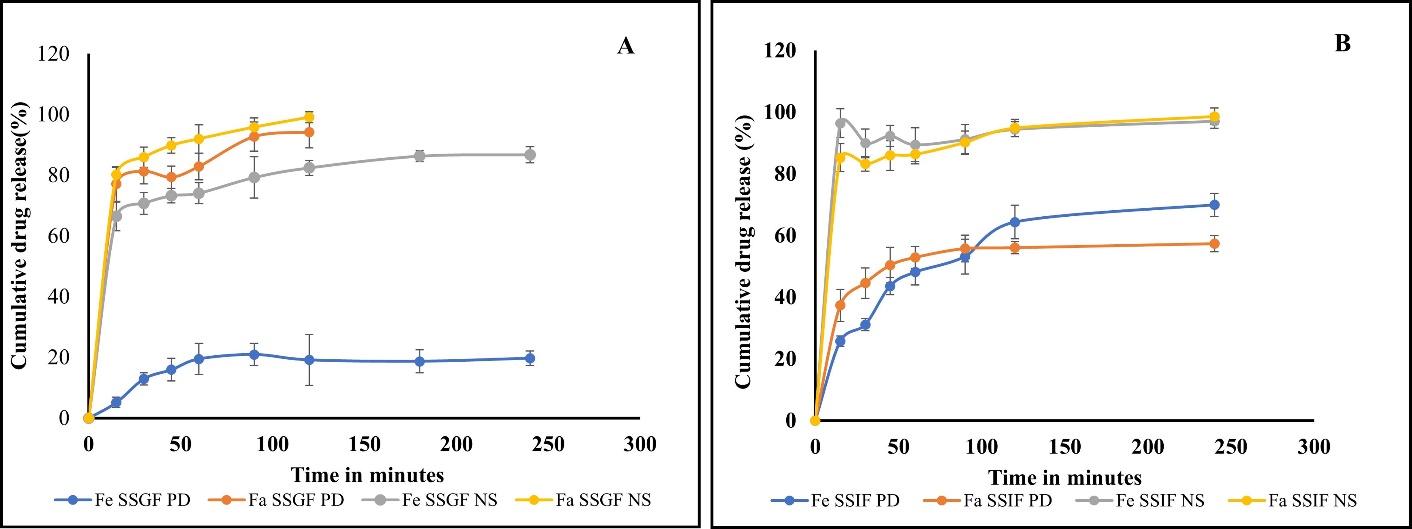
Fig. 5: In vitro drug release profiles of pure drug (PD) and NS in FaSSGF and FeSSGF media, B) Release profiles of IBR pure drug (PD) and NS in FaSSIF and FeSSIF (Mean±SD; n=3)
Stability studies
Three different temperature settings were used for ninety days to conduct stability testing on IBR-NS w. r. t PS. After the 60th day, the PS rose after remaining constant for the previous 60 days [25]. At a temperature of 2-8 °C, the size grew over 90 days, going from 83.22±12.89 nm to 88.860±8.99 nm. A decrease in stability over time is suggested by an increase size from 83.22±12.89 nm to 102.86±12.81 nm at 40 °C. The progressive rise in PS seen during stability testing was probably caused by Ostwald ripening, wherein small particles dissolve and re-precipitate onto larger particles. This is especially true at lower temperatures when the process may occur more slowly. The more pronounced increase at 40 °C indicates temperature's role in promoting agglomeration and aggregation. Temperature and storage conditions are two scenarios of variables that can affect crystal growth. The growth of crystals may have contributed to the increase in PS if the drug changed from an amorphous to a crystalline state during storage [26].
Pharmacokinetic studies
In both fasting and fed conditions, concentration-time profiles of the NS and PD were compared and the same is shown in fig. 6. The pharmacokinetics of the study is mentioned in table 2. Pharmacokinetic characteristics of the PD drug showed significant differences between fed and fasting circumstances, with decreased half-life after food intake and higher Cmax and AUC0-24h. In keeping with earlier formulation trials meant to lessen the effects of food, NS pharmacokinetic characteristics, however, did not significantly differ between fed and fasting settings. The formulation showed a 3.05-fold rise in AUC0-24h and a 3.97-fold rise in Cmax in comparison to the PD during fasting. According to earlier research, nanocrystals have improved adhesion, which could result in higher bioavailability. A delayed rate of stomach emptying, an improved blood supply, and the existence of bile salts and lipids, which promote medication solubilization at intestinal pH, could all contribute to the improved absorption in the fed state [27].
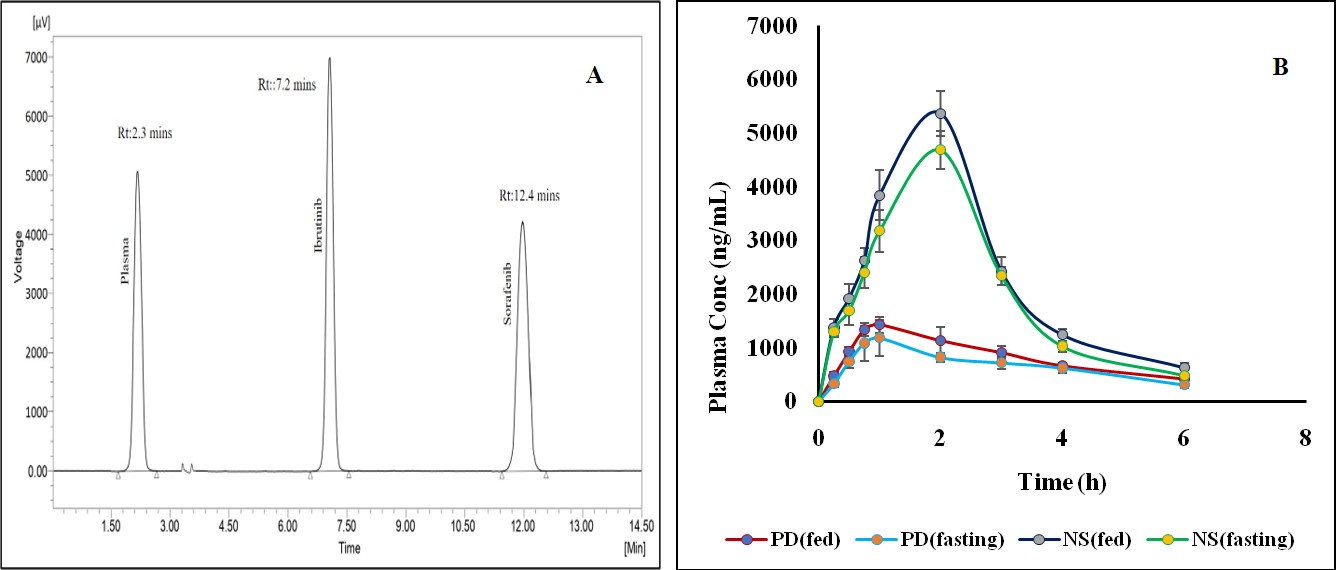
Fig. 6: Pharmacokinetic profiles in rats following oral administration
Table 2: Pharmacokinetic parameters
| Parameters | Plain drug (IBR) | Formulation (IBR NS) | ||
| Fast | Fed | Fast | Fed | |
| Cmax (ng/ml) | 1180.14±328.98 | 1426.9±142.89 | 4686.82±348.66 | 5370.46±414.66 |
| Tmax (h) | 1 | 1 | 2 | 2 |
| Half-life (h) | 2.73±0.77 | 2.73±0.54 | 1.21±0.97 | 1.31±0.61 |
| AUC0-t (ng. h/ml) | 4048.86±244.98 | 4048.86±186.34 | 12380.675±330.12 | 14173.858±329.26 |
| AUC0-∞ (ng. h/ml) | 5259.135±305.1 | 5259.135±281.53 | 13229.734±422.16 | 15369.251±458.02 |
| MRT(h) | 4.267±05.99 | 4.422±06.87 | 2.627±04.86 | 2.727±05.74 |
All the values were expressed in (n=3) Mean±SD
CONCLUSION
IBR NS, made with poloxamer 188 and HPMC E15, has PSs ranging from 134.6 to 214 nm. It was created utilizing the Box-Behnken design and HPH technique. Stability was verified using solid-state characterization, even during freeze-drying and a three-month storage period. Studies conducted in vitro revealed that NS released comparatively similar in both fasting and fed state of both gastric and intestinal contents than the standard pure medication. The NS showed an improved bioavailability and decreased variability between fed and fasting circumstances in rat pharmacokinetic studies, indicating that it may reduce pharmacokinetic variability.
ACKNOWLEDGMENT
The research facilities were provided by TRR College of Pharmacy, Hyderabad, for which the authors are grateful.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
S. S. C. and D. V. R. N. B planned the study and performed as per the protocol. V. V. R and S. R. P. performed the data analysis, whereas P. S and R. K. S guided the process to prepare the manuscript.
CONFLICT OF INTERESTS
The authors utter no conflict of interest.
REFERENCES
Aher SS, Malsane ST, Saudagar RB. Nanosuspension: an overview. Int J Curr Pharm Sci. 2017;9(3):19-23. doi: 10.22159/ijcpr.2017.v9i3.19584.
Masso Valles D, Jauset T, Soucek L. Ibrutinib repurposing: from B-cell malignancies to solid tumors. Oncoscience. 2016 May-Jun;3(5-6):147-8. doi: 10.18632/oncoscience.310, PMID 27489860.
Shakeel F, Iqbal M, Ezzeldin E. Bioavailability enhancement and pharmacokinetic profile of an anticancer drug ibrutinib by self nanoemulsifying drug delivery system. J Pharm Pharmacol. 2016 Jun;68(6):772-80. doi: 10.1111/jphp.12550, PMID 27018771.
De Jong J, Sukbuntherng J, Skee D, Murphy J, OBrien S, Byrd JC. The effect of food on the pharmacokinetics of oral ibrutinib in healthy participants and patients with chronic lymphocytic leukemia. Cancer Chemother Pharmacol. 2015 May;75(5):907-16. doi: 10.1007/s00280-015-2708-9, PMID 25724156.
Vidyadhari J, Gayatriramya M, Durga SP, Pavani P, Rajesh K. Nanosuspensions: a strategy to increase the solubility and bioavailability of poorly water soluble drugs. Asian J Pharm Clin Res. 2023 May;16(5):33-40. doi: 10.22159/ajpcr.2023.v16i5.46617.
Rangaraj N, Pailla SR, Chowta P, Sampathi S. Fabrication of Ibrutinib nanosuspension by quality by design approach: intended for enhanced oral bioavailability and diminished fast fed variability. AAPS PharmSciTech. 2019 Aug;20(8):326. doi: 10.1208/s12249-019-1524-7, PMID 31659558.
Rangaraj N, Pailla SR, Shah S, Prajapati S, Sampathi S. QbD aided development of ibrutinib loaded nanostructured lipid carriers aimed for lymphatic targeting: evaluation using chylomicron flow blocking approach. Drug Deliv Transl Res. 2020 Oct;10(5):1476-94. doi: 10.1007/s13346-020-00803-7, PMID 32519202.
Sowmya C, Suriyaprakaash KK, Abrar AH. Solid lipid nanoparticles: modern progress in nose to brain transduction. Int J Appl Pharm. 2023 Apr;15(4):20-6. doi: 10.22159/ijap.2023v15i4.47897.
Gera S, Talluri S, Rangaraj N, Sampathi S. Formulation and evaluation of naringenin nanosuspensions for bioavailability enhancement. AAPS PharmSciTech. 2017 Dec;18(8):3151-62. doi: 10.1208/s12249-017-0790-5, PMID 28534300.
Khalifa NE, Nur AO, Osman ZA. Artemether loaded ethyl cellulose nano suspensions: effects of formulation variables physical stability and drug release profile. Int J Pharm Pharm Sci. 2017 Jun;9(6):90-6. doi: 10.22159/ijpps.2017v9i6.18321.
Reddy KS, Bhikshapathi D. Design and optimization of DPC-crosslinked HPβCD nanosponges for entrectinib oral delivery: formulation characterization and pharmacokinetic studies. Futur J Pharm Sci. 2024 Mar;10(1):101. doi: 10.1186/s43094-024-00680-8.
Palanati M, Bhikshapathi DV. Development characterization and evaluation of entrectinib nanosponges loaded tablets for oral delivery. Int J App Pharm. 2023 Nov-Dec;15(6):269-81. doi: 10.22159/ijap.2023v15i6.49022.
Laxmi BV, Bhikshapathi D, Sailaja Rao P. Optimization and enhancement of oral bioavailability of dabrafenib as nanobubbles using quality by design approach. PharmSci. 2025 Jan;31(1):74-88. doi: 10.34172/PS.2024.31.
Pailla SR, Talluri S, Rangaraj N, Ramavath R, Challa VS, Doijad N. Intranasal zotepine nanosuspension: intended for improved brain distribution in rats. Daru. 2019 Jun;27(2):541-56. doi: 10.1007/s40199-019-00281-4, PMID 31256410.
Sunitha S, Rakesh A, Sujatha Dodoala D, Vijaya K. Biodegradable polymeric nanocarriers for oral delivery of antiretroviral drug: pharmacokinetic and in vitro permeability studies. J Appl Pharm Sci. 2021 Apr;11(4):28-39. doi: 10.7324/JAPS.2021.110405.
Liu D, Pan H, He F, Wang X, Li J, Yang X. Effect of particle size on oral absorption of carvedilol nanosuspensions: in vitro and in vivo evaluation. Int J Nanomedicine. 2015 Oct;10:6425-34. doi: 10.2147/IJN.S87143, PMID 26508852.
Sahu BP, Das MK. Nanosuspension for enhancement of oral bioavailability of felodipine. Appl Nanosci. 2014 Jun;4(2):189-97. doi: 10.1007/s13204-012-0188-3.
Verma S, Gokhale R, Burgess DJ. A comparative study of top down and bottom up approaches for the preparation of micro/nanosuspensions. Int J Pharm. 2009 Sep;380(1-2):216-22. doi: 10.1016/j.ijpharm.2009.07.005, PMID 19596059.
Singh A, Neupane YR, Panda BP, Kohli K. Lipid Based nanoformulation of lycopene improves oral delivery: formulation optimization ex vivo assessment and its efficacy against breast cancer. J Microencapsul. 2017 Jul;34(4):416-29. doi: 10.1080/02652048.2017.1340355, PMID 28595495.
Wang Y, Ma Y, Zheng Y, Song J, Yang X, Bi C. In vitro and in vivo anticancer activity of a novel puerarin nanosuspension against colon cancer with high efficacy and low toxicity. Int J Pharm. 2013 Oct;441(1-2):728-35. doi: 10.1016/j.ijpharm.2012.10.021, PMID 23089583.
Chen Z, Zhai J, Liu X, Mao S, Zhang L, Rohani S. Solubility measurement and correlation of the form a of ibrutinib in organic solvents from 278.15 to 323.15 K. J Chem Thermodyn. 2016 Nov;103:342-8. doi: 10.1016/j.jct.2016.08.034.
Celebi N, Gulbag Pinar S. Optimization and evaluation of cyclosporine a nanosuspension stabilized by combination stabilizers using high pressure homogenization method. Sanat. 2019;23(6):1009-21. doi: 10.35333/jrp.2019.65.
Lalatsa A, Schatzlein AG, Mazza M, Le TB, Uchegbu IF. Amphiphilic poly (L-amino acids) new materials for drug delivery. J Control Release. 2012 Jul;161(2):523-36. doi: 10.1016/j.jconrel.2012.04.046, PMID 22613882.
Lee J, Lee SJ, Choi JY, Yoo JY, Ahn CH. Amphiphilic amino acid copolymers as stabilizers for the preparation of nanocrystal dispersion. Eur J Pharm Sci. 2005 Jul;24(5):441-9. doi: 10.1016/j.ejps.2004.12.010, PMID 15784334.
Jacob S, Nair AB, Shah J. Emerging role of nanosuspensions in drug delivery systems. Biomater Res. 2020 Apr;24:3. doi: 10.1186/s40824-020-0184-8, PMID 31969986.
Dizaj SM, Vazifehasl Zh, Salatin S, Adibkia Kh, Javadzadeh Y. Nanosizing of drugs: effect on dissolution rate. Res Pharm Sci. 2015 Jun;10(2):95-108. doi: 10.4103/1735-5362.155641, PMID 26487886.
Hao J, Gao Y, Zhao J, Zhang J, Li Q, Zhao Z. Preparation and optimization of resveratrol nanosuspensions by antisolvent precipitation using box-behnken design. AAPS PharmSciTech. 2015;16(1):118-28. doi: 10.1208/s12249-014-0211-y, PMID 25209687.