
Int J App Pharm, Vol 17, Issue 6, 2025, 325-334Original Article
DEVELOPMENT AND VALIDATION OF A STABILITY-INDICATING HPTLC METHOD FOR VERAPAMIL ANALYSIS USING THE AQBD APPROACH
GAYATRI S. BADGUJAR, DHRUVI PRAJAPATI*
Department of Quality Assurance, School of Pharmacy, Parul University, P. O. Limda, Ta. Waghodia, Vadodara, Gujarat-391760, India
*Corresponding author: Dhruvi Prajapati; *Email: dap2611@gmail.com
Received: 03 Apr 2025, Revised and Accepted: 26 Sep 2025
ABSTRACT
Objective: To develop selective, precise and accurate High Performance Thin Layer Chromatography (HPTLC) method for forced degradation study of Verapamil by implementing AQbD (Analytical quality by design) approach. To optimize chromatographic conditions and detection wavelength for efficient separation and quantification of Verapamil using AQbD. To perform a forced degradation study on Verapamil as per International Council for Harmonisation (ICH) guideline. To validate the developed HPTLC method as per ICH Q2(R2) guideline.
Methods: The method employs TLC aluminium plates precoated with silica gel 60F254 as the stationary phase. The mobile phase consists of toluene, ethyl acetate, methanol, and ammonia in a volumetric ratio of 4:2:3.5:0.5. Verapamil quantification was performed in reflective mode at a wavelength of 276 nm. The method was validated for linearity, precision, accuracy, recovery, and robustness.
Results: The HPTLC method exhibited strong retention for verapamil with an Rf value of 0.72. Calibration curve analysis demonstrated excellent linearity over a concentration range of 200-1000 ng/band with a correlation coefficient (R²) of 0.9961. The slope and intercept were determined as 0.0013x+0.0074. The detection and quantitation limits were found to be 0.3008 ng/band and 0.9116 ng/band, respectively. The method showed high precision, accuracy, and robustness, ensuring its reliability for pharmaceutical applications.
Conclusion: The validated HPTLC method provides an efficient and reliable approach for the quantitative analysis of verapamil in synthetic mixtures. Its precision, sensitivity, and rapid analytical capabilities make it a suitable technique for pharmaceutical quality control and routine analysis.
Keywords: HPTLC, Verapamil, Stability-indicating method, Analytical quality by design (AQbD), Method validation
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i6.54435 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Verapamil (benzeneacetonitrile,-[3-[[2-(3,4-dimethoxyphenyl)-ethyl][methylamino] propyl]-3,4-dimethoxy-~-(1-methylethyl)-, monohydrochloride is a phenylalkylamine class calcium channel blocker [1]. Verapamil is water-soluble, very soluble in acidic solutions, easily soluble in chloroform, slightly soluble in alcohol, and not at all soluble in ether [2].

Fig. 1: Structure of verapamil
It also improves blood flow in the heart due to its calcium-blocking effect on the smooth muscles, it is responsible for anti-ischemic and anti-anginal efficacy in both reversible and irreversible types of coronary artery diseases and also as an antihypertensive, anti-angina, hypertrophic cardiomyopathy, and anti-arrhythmic [3-7]. Verapamil inhibits L-type calcium channels present in arteries' and heart's smooth muscle cells. Under normal circumstances, calcium enters these cells to cause muscular contraction. Verapamil blocks the entry of calcium, and thus smooth muscle relaxation and vasodilation (widening of arteries) [8].
Verapamil has been evaluated employing various analytical techniques, such as UV-visible spectrophotometric techniques, RP-HPLC techniques, Ultra Performance Liquid Chromatography (UPLC), and hyphenated techniques, as per the literature review. Nevertheless, no literature reports mention the HPTLC method for the estimation of verapamil [9-21]. Most of the pharmaceutical companies nowadays accept HPTLC as an analytical drug tool for quality control of pharmaceutical dosage forms. For being in accordance with the upcoming ICH Q1A(R2), Q2(R2), Q8(R2), Q9, and Q14 guidelines [22-25]. The aim of the current work is for developing with validating a stability-indicating HPTLC method for quantitative determination of Verapamil according to the Analytical Quality by Design (AQbD) approach. HPTLC method was established through the application of chemometric-based principal component analysis for the identification of high-risk method parameters [26, 27].
HPTLC is an advanced form of planar kind of chromatography which has become increasingly popular in recent years due to its wide range of applications. These include medicinal plant identification and evaluation, phytochemical analysis, therapeutic assessment, product and lichen ingredient testing, and detecting adulterants in formulations. HPTLC facilitates the rapid analysis of numerous samples and enhances sensitivity with maximum utilization of thin-layer chromatography with the least solvent volume. The objective of the current research is to develop a method for identifying Verapamil among its degradation products that is sensitive, precise, reproducible, and stable, using an Analytical Quality by Design (AQbD) strategy as per ICH guidelines. AQbD applies the Quality by Design (QbD) principle to construct analytical procedures so that quality is embedded in the process and not just evaluated in the final product. The Quality Target Method Profile (QTMP) or the Analytical Target Profile (ATP) is the first step of this method. It is used to direct decision-making in research and development. Additionally, stress testing or forced degradation studies are significant in drug research as they subject therapeutic ingredients or products to severe environmental conditions, triggering chemical and physical alterations. These studies help in the detection of likely degradation products and processes, ultimately resulting in a trustworthy and consistent analytical method.
MATERIALS AND METHODS
Instruments
The research was conducted with the HPTLC CAMAG TLC instrument (CAMAG, Muttenz, Switzerland). The samples were deposited as bands onto Merck TLC plates pre-coated with silica gel 60 F254 (20×10 cm, 0.2 mm thickness) using a Camag Linomat V applicator, employing a 100 ml sample syringe (Hamilton, Switzerland). Plates were pre-conditioned with methanol and heated at 60ºC for 10 min prior to sampling. At 25 ºC, the mobile phase of the chamber attained its optimum saturation time of 30 min. The chromatogram produced was 7 cm in length. The TLC plate was then dried using an air dryer. The slit dimensions were 6.0 mm by 0.45 mm, and it was scanning at a speed of 30 mm/s. Densitometric scanning was performed at 276 nm with WINCATS software (version 1.3.0) on a Camag Densitogram HPTLC scanner III.
Reagents and materials
The active pharmaceutical ingredient (API) of Verapamil was sourced from Surat, Gujarat, India, at Balaji Drugs. A TLC aluminium sheet, pre-coated with a silica gel layer of 60 F254, was utilized. Laboratory reagent (LR) quality toluene, ethyl acetate, and an ammonia solution were provided by Aatur Instrument Chemicals at Vadodara, Gujarat, India. The laboratory reagent (LR) grade methanol was supplied by Sulab Laboratory at Vadodara, Gujarat, India.
Preparation of standard stock solution
To prepare a stock standard solution, 10 mg of VER was dissolved in 10 ml of methanol, resulting in a concentration (1000 µg/ml). To obtain a 100 µg/ml solution, 1 ml of the stock solution was transferred to a volumetric flask of 10 ml and diluted to the mark with methanol. The prepared sample was kept at room temperature until required for plate application.
Selection of mobile phase
Several solvent solutions were tried as mobile phase, and Toluene: Methanol: Ammonia: Ethyl Acetate (4:3.5:0.5:2% v/v/v/v) was selected since it passed all of the system suitable acceptance requirements.
A range of solvent systems was evaluated to identify a suitable mobile phase for the effective HPTLC separation of Verapamil. Initial binary combinations such as methanol: ethyl ether, methanol: acetic acid, diethyl ether: methanol, hexane: isobutyl alcohol, and methanol: acetonitrile in various ratios failed to produce visible spots, indicating poor interaction or migration of the analyte. Toluene-based systems showed improved performance; toluene: acetonitrile (7:3) and toluene: ethyl acetate (7:3) yielded Rf values of 0.2 and 0.125, respectively, while acetonitrile: phosphate buffer (pH 2.2) showed no migration. Ternary systems such as toluene: acetone: glacial acetic acid (7:1:2), and those with ethyl acetate or acetonitrile and glacial acetic acid gave low Rf values around 0.07. Broader and tailing peaks were observed with combinations like toluene: acetonitrile: ethyl acetate (8:1:1) and toluene: ethyl acetate: methanol (7:2:1), showing Rf values of 0.3 and 0.295, respectively. Quaternary systems provided the best results; toluene: ethyl acetate: methanol: ammonia 4:2:3.5:0.5 with an Rf of 0.72. Thus, toluene-based quaternary solvent systems with ammonia or formic acid are most effective for Verapamil separation.
Selection of wavelength
The developed plate was scanned in spectrum mode by the Densitogram CD-60 TLC scanner and CAMAG software at the suitable Rf value for the location, and resultant spectrums were combined. Verapamil's multiwavelength Densitogram depicted maximum absorption at the 200-400 nm region. Maximum wavelength of 276 nm was employed in this study.
HPTLC method development
Chromatographic conditions
15 samples were spotted in 6 mm wide bands on precoated silica gel aluminium plates 60F with CAMAG microliter syringes and a CAMAG Linomat 5 sample applicator (CAMAG, Switzerland). Toluene, ethyl acetate, methanol, and ammonia were blended in the mobile phase in the following proportion: 4:2:3.5:0.5. Mobile phase was prepared using trial-and-error method, and chamber saturation time was 30 min, and the development went up to 80 mm length. The plates were dried in an air dryer. The plates were scanned with CAMAG TLC scanner 3 at 276 nm using win CATS software version 1.3.0. Development of method using AQbD approach.
Identification of QTMP, CMA, risk assessment
The initial step consisted of the establishment of the goals for developing the ATP method. The key objectives are the optimization of chromatographic conditions to enhance the quality of the chromatogram. The application of analytical quality by design (AQBD) in high-performance thin-layer chromatography (HPTLC) ensures a systematic and risk-based strategy to the development of a reliable and rugged method. In this work, central composite design (CCD) was employed for optimization of key chromatographic conditions. Analytical target profile (ATP) has been established to ensure accuracy, precision, resolution, robustness and sensitivity. Critical Analytic characteristics (CAAs) like delay factor (Rf) and peak surface (R²) were determined, and a risk grading with the aid of Ishikawa diagrams (fig. 2) and failure mode and effect analysis assistance identifies the critical method of methanol volume fitting and saturation time. A quality goal method profile (QTMP) and critical method-features (CMAs) were drawn up based on literature reviews and initial studies. Two-level CCD experimental design was used with levels (-1, 0,+1) to examine the effect of CMPs on CAAs (table 1).
Standard and sample were workable on pre-coated silica gel 60 F254 plates, which were developed in a saturated twin-trough chamber and scanned with a densitometric scanner. The data collected were analysed through Response Surface Methodology (RSM) to build a quadratic polynomial model, and contour plots and response surface plots were plotted to view interactions and optimize conditions. Robustness was assessed based on a risk-based approach adhering to ICH Q8 and Q9 guidelines. Validation of the optimized method according to ICH Q2 (R1) guidelines was done to prove accuracy, precision, linearity, and specificity. The incorporation of AQbD into HPTLC method development increases method reliability, regulatory compliance, and analytical consistency for routine analysis.

Fig. 2: Ishikawa (Fishbone) diagram representing critical factors affecting HPTLC performance
Table 1: Levels of critical method parameters used in central composite design (CCD) for optimization of HPTLC conditions
| Factor | Independent variables | Factor level low (-1) | Factor level medium (0) | Factor level high (+1) |
| A | Volume of methanol (ml) | 3 | 3.5 | 4 |
| B | Saturation time (min) | 25 | 30 | 35 |
This table presents the low, medium, and high levels of two independent variables-volume of methanol and saturation time-selected for CCD-based experimental design. These variables were chosen based on their critical impact on chromatographic response (Rf value), as part of the AQbD strategy for optimizing the HPTLC method for Verapamil.
The CCD design matrix includes a mixture of farm points, axial points and central points, placed in such a way to investigate the response surface in multiple directions, making it possible to assess both linear and square effects of the factors, along with the estimation of the curvature and the interaction effects. The CCD design matrix is shown in table 2.
Central composite design runs
The CCD matrix includes combinations of saturation time and methanol volume across factorial, axial, and centre points. These design points were selected to evaluate the individual and interactive effects of these variables on the Rf value, supporting robust optimization of method parameters using AQbD principles.
Table 2: Central composite design (CCD) matrix showing experimental runs for optimization of Rf value in verapamil HPTLC method
| S. No. | Pattern | Saturation time (min) | Methanol volume (ml) | Retention factor values |
| 1 | a0 | 25 | 3.5 | 0.67 |
| 2 | -+ | 25 | 4 | 0.69 |
| 3 | 00 | 30 | 3.5 | 0.72 |
| 4 | +- | 35 | 3 | 0.75 |
| 5 | 00 | 30 | 3.5 | 0.72 |
| 6 | -- | 25 | 3 | 0.79 |
| 7 | 0A | 30 | 4 | 0.75 |
| 8 | ++ | 35 | 4 | 0.78 |
| 9 | 0a | 30 | 3 | 0.67 |
| 10 | A0 | 35 | 3.5 | 0.72 |
In the CCD matrix, the coded patterns represent the levels of two variables: saturation time (25, 30, 35 min) and volume of methanol (3, 3.5, 4 ml), with "+" for high, "−" for low, "0" for centre, and "a/A" as axial points to evaluate curvature. The Rf value served as the response variable. Centre points (runs 3, 5, and 10) with 30 min and 3.5 ml showed consistent Rf values (~0.72), indicating method repeatability. Axial points (runs 1, 9, and 10) helped detect non-linearity. The highest Rf (0.79) was observed at the lowest factor levels (25 min, 3 ml) in run 6, while the combination of high saturation and volume of methanol (run 8) also showed a high Rf (0.78). These observations suggest that both variables significantly affect analyte migration and are essential for method optimization.
Validation of HPTLC method
System suitability
600 ng/band of VER was employed. The band specificity was assessed through spectrum measurement analysis. Wavelength 276 nm was used to measure the peak purity of VER through the spectra taken at Three distinct points were identified on the band: the start of the peak (S), the peak's apex (M), and the end of the peak (E).
Linearity
The linearity of the method was evaluated by analysing 2, 4, 6, 8, and 10 µl** of a working standard solution containing 100 µg/ml of Verapamil. The linear response was examined within the concentration range of 200-1000 ng/band. Each sample solution was analysed in triplicate, and the average peak area of Verapamil was calculated. A calibration curve was constructed by plotting the peak area against the corresponding concentration, and the correlation coefficient for Verapamil was determined.
Precision
Repeatability was evaluated by six determinations of the same concentration of VER at 600 ng/band. Data were recorded for every concentration, enabling the calculation of mean area, standard deviation (SD), and relative standard deviation (% RSD).
Intra-day precision was determined by taking different VER concentrations (200, 400, 600, 800, and 1000 ng/band) three times during one day.
Inter-day precision was checked by analyzing VER levels of 200, 400, 600, 800, and 1000 ng/band on three different days.
Recovery
Procedure accuracy was confirmed by spiking a drug standard solution in the already analyzed sample solution. The recovery check was carried out at three different levels: 80%, 100%, and 120%. The experiment was repeated three times.
Specificity
Stress degradation study of VER by HPTLC. In line with ICH recommendations, stress tests were performed (QIA) (R2). The Verapamil underwent multiple stress scenarios, such as hydrolysis, photolysis, oxidation, and both acidic and alkaline stress.
Stability Indicating Study of VER for HPTLC
According to ICH guidelines, stability tests were conducted (QIA) (R2). The VER was subjected to various stress conditions like hydrolysis, photolysis, oxidation, and acidic as well as alkaline stress.
Acidic stress degradation
1 ml of the standard VER solution was mixed with 1 ml of 0.1 M HCl in a 10 ml volumetric flask and refluxed at 70 °C for 60 min. After cooling, it was neutralized with 1 ml 0.1 N NaOH and diluted to volume with methanol. Then, 1 ml of this solution was taken and further diluted with 9 ml of methanol in a 10 ml Volumetric Flask, and the chromatography was checked under optimized conditions.
Alkaline stress degradation
1 ml of the standard VER solution was mixed with 1 ml of 0.1 N NaOH in a 10 ml volumetric flask and refluxed at 70 °C for 60 min. After cooling, it was neutralized with 1 ml 0.1 M HCL and diluted to volume with methanol. Then, 1 ml of this solution was taken and further diluted with 9 ml of methanol in a 10 ml Volumetric Flask, and the chromatography was checked under optimized conditions.
Oxidative stress degradation
For the degradation under oxidative stress, 10 mg of VER was accurately measured and placed into a 10 ml volumetric flask, followed by the addition of 1 ml of 3 % H₂O₂. The resulting mixture was then kept in a water bath at a temperature of 70 °C for 60 min. After this period, 1 ml of the solution was transferred to a 10 ml volumetric flask and adjusted to the final volume with methanol.
Thermal stress degradation
Degradation under Thermal stress was conducted by spreading 50 mg of VER as a thin layer in a petri dish and placing it in a microwave oven for 8 h. Then, 10 mg of VER was dissolved in methanol, and the total volume was adjusted to 10 ml in a volumetric flask.
Photolytic stress degradation
Degradation under photolytic stress was conducted by spreading 50 mg of VER as a thin layer in a petri dish and placing it in a UV chamber with a wavelength of 276 nm for 14 h. Following this, 10 mg of VER was dissolved in methanol, and the total volume was adjusted to 10 ml in a volumetric flask.
Robustness
For the robustness test, minor adjustments were applied to the composition of the mobile phase, and the impact on the results was examined. Various Toluene: Ethyl acetate: Methanol: Ammonia (4: 2: 3.5: 0.5 v/v/v/) ratios were chosen A (4: 2.5: 3: 0.5 v/v/v/v), B (4: 1.5: 4: 0.5 v/v/v/v) and chromatograms were run while changing the saturation time (30±5 min, having values of 25, 30, or 35 min).
Preparation of synthetic mixture
A synthetic blend was prepared by precisely weighing VER (API) and blending it with the pharmaceutically employed excipients like microcrystalline cellulose, magnesium stearate, talc etc. These excipients were selected to mimic the composition of commercial formulations to provide a realistic representation of the real dosage form. The ingredients were well mixed with a mortar and pestle to achieve uniform distribution of the drug. The resultant mixture was stored in an airtight container at room temperature for subsequent analysis.
RESULTS AND DISCUSSION
A Central Composite Design (CCD) was employed to evaluate the influence of two critical method parameters-saturation time (A) and volume of methanol (B)-on the retardation factor (Rf) in HPTLC method development. The analysis aimed to optimize these parameters for improved chromatographic performance.


(A) (B)
Fig. 3: (A) Contour plot representing the effect of saturation time and methanol volume on Rf Value. (B) 3D surface plot depicting the interaction between saturation time and methanol volume on retention factor value
Contour plot analysis
The contour plot (fig. 3-A) illustrates the interaction effects between saturation time and methanol volume on the Rf value. The plot indicates a well-defined region where the desired Rf value is achieved, marked by the green zone. The optimal conditions identified reflect minimal variability in the central region, confirming stable chromatographic performance.
3D surface plot analysis
The 3D surface plot (fig. 3-B) visualizes the influence of both factors on the Rf value. A clear peak at the centre highlights the optimal conditions for achieving the target Rf value. The upward slope indicates a direct relationship between saturation time and Rf, while the volume of methanol shows a comparatively moderate effect.


(A) (B)
Fig. 4: (A) Normal probability plot of residuals for retention factor value model (B) ANOVA table for retention factor value model
Residual analysis
The normal plot of residuals (fig. 4-A) demonstrates that data points align closely with the regression line, confirming the model’s adequacy and normal distribution of residuals. This suggests that no significant outliers are present, validating the consistency of the experimental data.
ANOVA analysis
The ANOVA table (fig. 4-B) confirms the statistical significance of the model with a p-value of 0.0428 (<0.05), indicating that the selected factors have a meaningful impact on the Rf value. Of the two parameters studied, saturation time exhibited a significant effect (p = 0.0159), whereas mobile phase composition was found to be statistically insignificant (p = 0.6542). Additionally, the non-significant lack of fit (p = 0.7751) supports the adequacy and reliability of the model for predicting Rf values. Despite its insignificance, mobile phase composition was retained in the CCD model due to its crucial role in influencing chromatographic behaviour, including peak shape, resolution, and retention time. Its inclusion also allowed for assessment of potential interactions and helped maintain the robustness of the model, aligning with best practices in analytical Quality by Design (QbD).
The integration of CCD successfully optimized the HPTLC method by identifying optimal saturation time and volume of methanol for achieving the desired Rf value. The developed model proved statistically significant, with saturation time emerging as the key influencing factor. The method is robust, reproducible, and suitable for accurate analysis in future studies.
Method validation results
Linearity
Linearity in the range of 200-1000 ng/band was observed for VER. Linearity data are presented in the provided table 3. Single lane chromatogram, UV spectra and 3D overlain chromatogram are represented in fig. 5.
Table 3: Linearity data for VER (n = 3)
| Concentration (ng/band) | Mean peak area±SD (n = 3) | % RSD |
| 200 | 0.00975±0.00010 | 1.037 |
| 400 | 0.01250±0.00015 | 1.275 |
| 600 | 0.0154±0.00015 | 1.030 |
| 800 | 0.01755±0.00013 | 0.773 |
| 1000 | 0.01983±0.00012 | 0.613 |
Value expressed as mean±SD (n = 3)


A B

C
Fig. 5: (A) Overlay UV Absorption Spectra of VER (B) Calibration curve of mean peak area vs concentration (C) HPTLC Densitogram of VER
Precision
Repeatability
Repeatability study was performed with the use of VER solution 600 ng/band and SD and % RSD was found to be 1.54 (table 4)
Intermediate precision
In inter-day precision % RSD was found to be 0.88 and in intra-day precision, % RSD was found to be 0.8 (table 5 and 6 respectively). The inter-day and intra-day precision was found to similar, the close agreement between intra-day and inter-day precision results demonstrates that the analytical method is reproducible and exhibits good intermediate precision, confirming its reliability for routine analysis.
Accuracy
The quantity of VER was determined, and the percentage recovery is presented in table 7. As illustrated in table 7, the proposed method achieved a recovery rate of 98% and 120% across the three evaluated levels for 3 times by spiking the synthesis mixture (placebo) (80%, 100%, and 120%).
Limit of detection (LOD) and limit of quantification (LOQ)
LOD was calculated with the formula:
LOD = 3.3 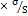
LOQ was calculated with the formula:
LOQ = 10
Where:  = Standard deviation of VER S = Slope of the calibration curve.
= Standard deviation of VER S = Slope of the calibration curve.
System suitability
System suitability was perform using 600 ng/band concentration six time % RSD was calculated (table 9) Peak purity data as mentioned in table 10.
r(s,m): correlation coefficient from start point to middle
All values of r(s,m) are above 0.999, and r(e,m) values are above 0.996. According to widely accepted HPTLC method validation criteria, a peak is considered spectrally pure if both correlation values are ≥ 0.990. This ensures that the peak represents a single component and is not overlapped or contaminated by adjacent analytes or degradation products.
Thus, the data justifies the spectral purity of verapamil peaks, supporting the specificity and reliability of the analytical method for routine quality control and stability studies.
Robustness
The impact of slight variations in volume of methanol (±0.5 ml) and chamber saturation time (± 5 min) was evaluated. The outcomes of these modifications on the Rf value and peak area are presented in table 11.
Table 4: Repeatability data for VER (n = 6)
| Concentration (ng/band) | Peak area | Mean peak area±SD (n=6) | % RSD |
| 600 | 0.01565 | 0.01554±0.00024 | 1.54 |
| 0.01576 | |||
| 0.01582 | |||
| 0.01551 | |||
| 0.01530 | |||
| 0.01524 |
Value expressed as mean±SD (n = 6)
Table 5: Inter-day precision data for VER (n = 3 per level)
| Concentration (ng/band) | Mean peak area±SD (n = 3) | % RSD |
| 200 | 0.00967±0.000117 | 1.18 |
| 400 | 0.01255±0.000156 | 1.24 |
| 600 | 0.01548±0.000121 | 0.78 |
| 800 | 0.01754±0.000105 | 0.59 |
| 1000 | 0.01976±0.000129 | 0.65 |
Value expressed as mean±SD (n = 3)
Table 6: Intra-day precision data for VER (n = 3 per level)
| Concentration (ng/band) | Mean peak area±SD (n = 3) | % RSD |
| 200 | 0.009823±0.000117 | 1.18 |
| 400 | 0.01255±0.000156 | 1.24 |
| 600 | 0.01562±0.000121 | 0.78 |
| 800 | 0.01765±0.000105 | 0.59 |
| 1000 | 0.01992±0.000129 | 0.65 |
| mean % Relative Standard Deviation: 0.88 |
Value expressed as mean±SD (n = 3)
Table 7: Recovery data for VER (n = 3 per level)
| S. No. | Amount taken (ng/band) | Concentration level (n=3) | Amount drug added (ng/band) | Amount recovered (ng/band) | % Recovery±SD |
| 1 | 400 | 80% | 320 | 708.56 | 98.92±1.25 |
| 2 | 400 | 80% | 320 | 708.72 | |
| 3 | 400 | 80% | 320 | 709.06 | |
| 4 | 400 | 100% | 400 | 798.26 | 101.56±1.05 |
| 5 | 400 | 100% | 400 | 798.93 | |
| 6 | 400 | 100% | 400 | 799.12 | |
| 7 | 400 | 120% | 480 | 875.92 | 100.81±0.46 |
| 8 | 400 | 120% | 480 | 876.35 | |
| 9 | 400 | 120% | 480 | 876.88 |
Table 8: LOD and LOQ data for VER
| Limit of detection | 0.30083 (ng/band) |
| Limit of quantification | 0.91160 (ng/band) |
Table 9: System suitability data for VER (n=6)
| Track number | Retention factor value | Mean peak area±SD (n=6) | % RSD |
| 1 | 0.719 | 0.719±0.000894 | 0.1243 |
| 2 | 0.718 | ||
| 3 | 0.718 | ||
| 4 | 0.719 | ||
| 5 | 0.72 | ||
| 6 | 0.72 |
Value expressed as mean±SD (n = 6)
Table 10: Peak purity data for VER (n=6)
| Track number | Retention factor value | r(s,m) | r(e,m) |
| 1 | 0.719 | 0.999476 | 0.996897 |
| 2 | 0.718 | 0.999455 | 0.996484 |
| 3 | 0.718 | 0.999294 | 0.996733 |
| 4 | 0.719 | 0.999538 | 0.996037 |
| 5 | 0.72 | 0.999264 | 0.996037 |
| 6 | 0.72 | 0.999491 | 0.997012 |
Table 11: Robustness data for VER (n=3)
| Factor | Condition | Mean peak area±SD (n=3) | % RSD |
| Methanol volume (ml) | 3 | 0.01123±0.00015 | 1.3117 |
| 3.5 | 0.00965±0.00016 | 1.6677 | |
| 4 | 0.01221±0.0017 | 1.4137 | |
| Saturation time (min) | 25 | 0.015767±0.000204 | 1.295 |
| 30 | 0.0164±0.0002 | 1.219 | |
| 35 | 0.012523±0.00018 | 1.440 | |
| Value expressed as mean±SD (n = 3) |
Forced degradation study
The forced degradation study proves that VER degrades extensively in acidic, alkaline, oxidative and thermal conditions but is stable in photolytic conditions. The HPTLC method developed proved to effectively separate VER standard active pharmaceutical ingriedents (STD API) from its degradants (DP), proving its stability-indicating nature (fig. 6-10). Retention factor (Rf), peak purity, and area under the curve (AUC) were checked for all stress conditions to prove method specificity (table 12).

Fig. 6: VER and its degradation products under acidic stress conditions

Fig. 7: VER and its degradation products under alkaline stress conditions

Fig. 8: VER and its degradation products under oxidative stress conditions

Fig. 9: VER and its degradation products under thermal stress conditions

Fig. 10: VER and its degradation products under photolytic stress conditions
Table 12: Degradation study data of VER
| Stress conditions | % Active ingredient remain | % Product degraded |
| Acidic stress condition | 29 % | 71 % |
| Alkaline stress condition | 47.11 % | 52.89 % |
| Oxidative stress condition | 45 % | 55 % |
| UV light stress condition | 85 % | 15 % |
| Photolytic stress condition | 78 % | 22 % |
Forced degradation study revealed that VER experienced significant degradation under acidic, alkaline, oxidative, thermal and photolytic stress conditions. Percentage of degradation was calculated by comparing the area of the degraded sample with the sample solution, which was not degraded (100% pure API). Area of acidic stress, alkaline stress, oxidative stress, UV light stress, photolytic stress was found to be 0.00626, 0.00466, 0.00484, 0.00132, 0.00193, respectively. The area of the without degraded sample was found to be 0.008816.
Verapamil, a calcium channel blocker with ester, amine, and aromatic methoxy groups, showed considerable degradation under acidic, alkaline, oxidative, and thermal conditions because of the intrinsic instability of these functional groups. Under acidic conditions, the hydrolysis of the ester linkage was catalyzed by acid via the protonation of the ester oxygen, which increased its vulnerability to nucleophilic attack, leading to a degradation of 71%. In alkaline conditions, the hydroxide ion's attack during base-catalyzed hydrolysis severed the ester bond, resulting in a 52.89% degradation. Oxidative stress from hydrogen peroxide facilitated the N-oxidation of the tertiary amine and the hydroxylation of methoxy-substituted aromatic rings, resulting in 55% degradation. Thermal stress resulted in moderate deterioration (15–22% degradation) from heat-induced rearrangements or bond cleavage, especially in ester or methoxy groups. Conversely, photolytic stress at 276 nm caused approximately 15–22% degradation, suggesting that verapamil is quite photostable, probably because it lacks highly photoreactive groups. These findings validate that the established HPTLC technique successfully distinguishes verapamil from its degradation products, demonstrating its capacity to indicate stability.
The percent of degradation was calculated by using the given formula and results are mention in the table 12.
Calculation for the degraded product (% DP):  × 100
× 100
Table 13: Validation parameter summary
| S. No. | Parameters | Result |
| 1 | Range (ng/band) | 200 – 1000 |
| 2 | Correlation Coefficient | R² = 0.9961 |
| 3 | % Recovery | 98.92 – 101.56 % |
| 4 | Repeatability | 1.5414 % RSD |
| 5 | Inter-day precision | 0.59 – 1.24 % RSD |
| 6 | Intra-day precision | 0.59 – 1.24 % RSD |
| 7 | System suitability | 0.1243 % RSD |
| 8 | LOD (ng/band) | 0.30083 |
| 9 | LOQ (ng/band) | 0.911606 |
DISCUSSION
The present study successfully developed and validated a stability-indicating HPTLC method for the determination of Verapamil using the Analytical Quality by Design (AQbD) approach. Central Composite Design (CCD) optimization identified saturation time and methanol volume as critical parameters influencing chromatographic performance, with an optimal Rf value of 0.72 achieved under selected conditions. The robustness of the model, supported by low %RSD values across different runs, demonstrates that the method is reliable and reproducible under minor procedural variations, aligning with ICH Q2(R2) validation guidelines.
Forced degradation studies revealed that Verapamil is highly susceptible to acid-catalyzed (71% degradation) and base-catalyzed (52.89% degradation) hydrolysis, which can be attributed to cleavage of its ester linkage under extreme pH conditions. This finding is consistent with previous HPLC-based stability studies, such as dos Santos Moreira and Lourenço (2020), who reported significant degradation of Verapamil in acidic and alkaline conditions [28, 29]. The oxidative degradation observed (55%) is also in agreement with Walles et al. (2002), who demonstrated that Verapamil undergoes oxidative metabolism through N-oxidation of tertiary amines and hydroxylation of aromatic rings [30]. Thermal stress caused moderate degradation (15–22%), while minimal photolytic degradation (15–22%) indicates relative photostability, in line with earlier chromatographic investigations [31].
When compared to previous analytical methods, the proposed HPTLC approach demonstrates several advantages. Kowalczuk (2005) reported a densitometric HPTLC method for Verapamil and Trandolapril, but without an AQbD framework [32]. Similarly, Vijayabaskar and Mahalingam (2017) employed UPLC for Verapamil analysis with a higher solvent requirement and longer analysis time [33]. In contrast, the present method integrates AQbD principles with CCD-based optimization, ensuring systematic identification and control of high-risk variables, thus improving robustness and reproducibility. Furthermore, the obtained linearity (200–1000 ng/band, R² = 0.9961), precision (intra-day %RSD = 0.59–1.24; inter-day %RSD = 0.59–1.24), and recovery (98.9–101.5%) compare favourably with RP-HPLC and LC-MS/MS methods reported in earlier studies [9–12, 16, 17]. Importantly, the sensitivity of this HPTLC method, with an LOD of 0.30 ng/band and LOQ of 0.91 ng/band, is superior to many reported HPLC/UPLC methods, which typically exhibit higher detection limits.
Another notable advantage is the environmental sustainability of the method. Compared to conventional HPLC and UPLC techniques, this HPTLC procedure consumes smaller solvent volumes, involves shorter analysis times, and provides comparable or better analytical performance. Such features align with the principles of green analytical chemistry, making this approach suitable for routine pharmaceutical quality control and stability testing.
Overall, the integration of AQbD principles with HPTLC provides a systematic, sensitive, and eco-friendly stability-indicating method for Verapamil. Its validated robustness, high specificity, and ability to separate drug and degradation products under a range of stress conditions highlight its applicability in pharmaceutical analysis.
CONCLUSION
The AQbD-based stability-indicating HPTLC method developed in this study provides a robust, precise, and specific approach for the estimation of Verapamil in the presence of its degradation products. CCD optimization enabled the identification and control of critical method variables, ensuring reliable performance under minor procedural variations. Forced degradation studies confirmed the susceptibility of Verapamil to acidic, alkaline, and oxidative conditions, while demonstrating relative stability under thermal and photolytic stress. The method met ICH Q2(R2) criteria for accuracy, precision, robustness, and specificity, and effectively separated the drug from its degradation products, establishing its suitability for routine quality control and stability testing. The shorter analysis time, lower solvent consumption, and integration of AQbD principles make this method a sustainable and efficient alternative to conventional chromatographic approaches for Verapamil analysis.
ABBREVIATIONS
HPTLC – High-Performance Thin Layer Chromatography, AQbD – Analytical Quality By Design, API – Active Pharmaceutical Ingredient, ICH – International Council for Harmonisation, QTMP – Quality Target Method Profile, ATP – Analytical Target Profile, CMA – Critical Method Attribute, CCD – Central Composite Design, Rf – Retention Factor, UV – Ultraviolet, LOD – Limit Of Detection, LOQ – Limit Of Quantification, R² – Correlation Coefficient, SD – Standard Deviation, % RSD – Percentage Relative Standard Deviation, LR – Laboratory Reagent, HCl – Hydrochloric Acid, H₂O₂ – Hydrogen Peroxide, CMPs – Critical Method Parameters, CAAs – Critical Analytical Attributes, RSM – Response Surface Methodology, AUC – Area Under The Curve, RP-HPLC – Reverse Phase High-Performance Liquid Chromatography, UPLC – Ultra-Performance Liquid Chromatography, MS/MS – Mass Spectrometry.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
CONFLICT OF INTERESTS
Declared none
REFERENCES
Sica DA, Prisant LM. Pharmacologic and therapeutic considerations in hypertension therapy with calcium channel blockers: focus on verapamil. The J of Clinical Hypertension. 2007 Feb;9(2):1-22. doi: 10.1111/j.1524-6175.2007.06504.x.
Moreira CS, Lourenco FR. Development and optimization of a stability-indicating chromatographic method for verapamil hydrochloride and its impurities in tablets using an analytical quality by design (AQbD) approach. Microchem J. 2020 May 1;154:104610. doi: 10.1016/j.microc.2020.104610.
Anavekar SN, Christophidis N, Louis WJ, Doyle AE. Verapamil in the treatment of hypertension. J Cardiovasc Pharmacol. 1981;3(2):287-92. doi: 10.1097/00005344-198103000-00007, PMID 6166800.
Brodsky SJ, Cutler SS, Weiner DA, Mc Cabe CH, Ryan TJ, Klein MD. Treatment of stable angina of effort with verapamil: a double-blind placebo-controlled randomized crossover study. Circulation. 1982 Sep;66(3):569-74. doi: 10.1161/01.cir.66.3.569, PMID 6807568.
Schamroth L, Krikler DM, Garrett C. Immediate effects of intravenous verapamil in cardiac arrhythmias. Br Med J. 1972 Mar 11;1(5801):660-2. doi: 10.1136/bmj.1.5801.660, PMID 5015294.
Lakshmi AP, Kumar GA, Reddy TK, Kumar MA. Development and in vitro evaluation of gastroretentive verapamil HCl floating tablets. Int J Pharm Pharm Sci. 2012;4(1):360-3.
Epstein SE, Rosing DR. Verapamil: its potential for causing serious complications in patients with hypertrophic cardiomyopathy. Circulation. 1981;64(3):437-41. doi: 10.1161/01.cir.64.3.437, PMID 7196300.
Walles M, Thum T, Levsen K, Borlak J. Verapamil: new insight into the molecular mechanism of drug oxidation in the human heart. J Chromatogr A. 2002;970(1-2):117-30. doi: 10.1016/s0021-9673(02)00641-6, PMID 12350087.
Trivedi L, Telrandhe R, Dhabarde D. Differential spectrophotometric method for estimation and validation of verapamil in tablet dosage form. Int J Pharm Drug Anal. 2017;5(11):419-22.
Bhaskar R, Bhaskar R, Sagar MK, Saini V, Bhat K. Simultaneous determination of verapamil hydrochloride and gliclazide in synthetic binary mixture and combined tablet preparation by chemometric assisted spectroscopy. JASMI. 2012;2(3):161-6. doi: 10.4236/jasmi.2012.23026.
Logoyda L. Bioanalytical method development and validation for the simultaneous determination of verapamil and enalapril in the presence of enalaprilat by HPLC-MS/MS. Int J Appl Pharm. 2018;10(3):19-27. doi: 10.22159/ijap.2018v10i3.23974.
Vusuvandla G. Development and validation of RP-HPLC method for the simultaneous estimation of verapamil hydrochloride and trandolapril in bulk and pharmaceutical dosage forms. Asian J Pharm Anal Med Chem. 2016;4(1):38-46.
Valvo L, Alimenti R, Alimonti S, Raimondi S, Foglietta F, Campana F. Development and validation of a liquid chromatographic method for the determination of related substances in verapamil hydrochloride. J Pharm Biomed Anal. 1997;15(7):989-96. doi: 10.1016/s0731-7085(96)01923-1, PMID 9160266.
Venkatesh G, Ramanathan S, Mansor SM, Nair NK, Sattar MA, Croft SL. Development and validation of RP-HPLC-UV method for simultaneous determination of Buparvaquone atenolol propranolol quinidine and verapamil: a tool for the standardization of rat in situ intestinal permeability studies. J Pharm Biomed Anal. 2007;43(4):1546-51. doi: 10.1016/j.jpba.2006.11.013, PMID 17157469.
Gumustas M, Sanlı S, Sanlı N, Ozkan S. Development and validation of a liquid chromatographic method for concurrent assay of weakly basic drug verapamil and amphoteric drug trandolapril in pharmaceutical formulations. J Food Drug Anal. 2012;20:588-96.
Yatha R, Rajkamal B. An improved LC-MS/MS method development and validation for the determination of trandolapril and verapamil in human. Int J Pharm Sci. 2019;11(3):91-5. doi: 10.22159/ijpps.2019v11i3.31247.
Hefnawy M, Al Majed A, Alrabiah H, Algrain N, Mohammed M, Jardan YB. Rapid and sensitive LC-MS/MS method for the enantioanalysis of verapamil in rat plasma using superficially porous silica isopropyl cyclofructan 6 chiral stationary phase after SPE: application to a stereoselective pharmacokinetic study. J Pharm Biomed Anal. 2021;201:114108. doi: 10.1016/j.jpba.2021.114108, PMID 33962179.
Vijayabaskar S, Mahalingam V, Kalaivani. Analytical method development and validation for the analysis of verapamil hydrochloride and its related substances by using ultra-performance liquid chromatography. J Pharm Biomed Anal. 2017;137:189-95. doi: 10.1016/j.jpba.2017.01.006, PMID 28131058.
Sharma S, Shrestha B, Bhuyan NR, Majumdar S, Chowdhury S, Mazumder R. Chemometric method development for the determination of naringin and verapamil. Bull Natl Res Cent. 2024 Jan 15;48(1):13. doi: 10.1186/s42269-024-01169-3.
Reddy KT, Haque MA. Bioanalytical method development and validation of atrasentan in human plasma using verapamil as internal standard by liquid chromatography coupled with tandem mass spectrometry. Int J Health Sci. 2022;6 Suppl 8:625-38. doi: 10.53730/ijhs.v6nS8.10470.
Kowalczuk D. Simultaneous high-performance thin-layer chromatography densitometric assay of trandolapril and verapamil in the combination preparation. J AOAC Int. 2005;88(5):1525-9. doi: 10.1093/jaoac/88.5.1525, PMID 16386004.
International Council for Harmonisation. ICH guideline Q1A(R2): stability testing of new drug substances and products. Geneva: ICH; 2003.
International Council for Harmonisation. ICH guideline Q2(R2): validation of analytical procedures. Geneva: ICH; 2003.
International Council for Harmonisation. ICH Q9. Quality risk management. Geneva: ICH; 2003.
International Council for Harmonisation. ICH Q14. Analytical procedure development. Geneva: ICH; 2003.
Kulkarni RN, Pandhare RB, Deshmukh VK, Mohite PB, Pawar AR. High-performance thin-layer chromatography: a powerful analytical technique in pharmaceutical drug discovery. J Pharm Biol Sci. 2021;9(1):1-7.
Jain A, Parashar AK, Nema RK, Narsinghani T. High-performance thin-layer chromatography (HPTLC): a modern analytical tool for chemical analysis. Curr Res PharmSci. 2014;4(1):8-14.
Lakshmi AP, Kumar GA, Reddy TK, Kumar MA. Development and in vitro evaluation of gastroretentive verapamil HCl floating tablets. Int J Pharm Pharm Sci. 2012;4(1):360-3.
Moreira CS, Lourenco FR. Development and optimization of a stability-indicating chromatographic method for verapamil hydrochloride and its impurities in tablets using an analytical quality by design (AQbD) approach. Microchem J. 2020 May 1;154:104610. doi: 10.1016/j.microc.2020.104610.
Walles M, Thum T, Levsen K, Borlak J. Verapamil: new insight into the molecular mechanism of drug oxidation in the human heart. J Chromatogr A. 2002 Sep 13;970(1-2):117-30. doi: 10.1016/s0021-9673(02)00641-6, PMID 12350087.
Vijayabaskar S, Mahalingam V, Kalaivani. Analytical method development and validation for the analysis of verapamil hydrochloride and its related substances by using ultra perfomance liquid chromatography. J Pharm Biomed Anal. 2017 Apr 15;137:189-95. doi: 10.1016/j.jpba.2017.01.006, PMID 28131058.
Kowalczuk D. Simultaneous high-performance thin-layer chromatography densitometric assay of trandolapril and verapamil in the combination preparation. J AOAC Int. 2005 Sep 1;88(5):1525-9. doi: 10.1093/jaoac/88.5.1525, PMID 16386004.
Gumustas M, Sanli S, Sanli N, OzkAN SA. Development and validation of a liquid chromatographic method for concurrent assay of weakly basic drug verapamil and amphoteric drug trandolapril in pharmaceutical formulations. J Food Drug Anal. 2012;20(3):14. doi: 10.6227/jfda.2012200304.