
Int J App Pharm, Vol 18, Issue 1, 2026, 464-478Original Article
DESIGN, SYNTHESIS, AND EVALUATION OF NOVEL TYROSINASE INHIBITORS FOR SKIN DEPIGMENTATION
AMANI Y. SALMAN1, MANAL M. NAJDAWI1,2*, MAHMOUD A. AL-SHA’ER3,8, NAWZAT D. AL JBOUR3,9, RAWAND M. DAGHMASH4, LOAY K. HASSOUNEH5,6, ZEAD H. ABUDAYEH1, ALA’ Y. SIRHAN7, MOHAMMED AL-AKEEDI10
1Department of Applied Pharmaceutical Sciences and Clinical Pharmacy, Faculty of Pharmacy, Isra University, Amman, Jordan. 2Faculty of Pharmacy, Department of pharmaceutical technology and cosmetics, Middle East University, Amman, Jordan. 3Department of Pharmaceutical Sciences Faculty of Pharmacy, Zarqa University, Zarqa-13110, Jordan. 4Department of Pharmaceutics and Pharmaceutical Technology, Faculty of Pharmacy, Al-Ahliyya Amman University, Amman, Jordan. 5Department Biology Education, Faculty of Education and Arts, Sohar University-311 Sohar, Sultanate of Oman. 6Department of Respiratory Therapy, Faculty of Allied Medical Sciences, Isra University, Amman, Jordan. 7Department of Pharmacy, Faculty of Pharmacy, Amman Arab University, Amman, Jordan. 8Pharmaceutical Research Center, Zarqa University, Zarqa-13110, Jordan, 9Department of Pharmaceutical Technology, Faculty of Pharmacy, Zarqa University, Zarqa-13110, Jordan, 10 College of Pharmacy, The University of Mashreq, Bagdad, Iraq
*Corresponding author: Manal M. Najdawi; *Email: m.najdawi@meu.edu.jo
Received: 06 Apr 2025, Revised and Accepted: 08 Nov 2025
ABSTRACT
Objective: Hyperpigmentation is a prevalent dermatological condition caused by excessive melanin production, primarily catalyzed by the enzyme tyrosinase. Existing depigmenting agents, such as hydroquinone and kojic acid, are limited by cytotoxicity, instability, and suboptimal efficacy. This study aimed to design, synthesize, and evaluate structurally novel tyrosinase inhibitors derived from cinnamic acid to overcome these limitations.
Methods: Tencinnamic acid-based derivatives were designed with strategic modifications, including esterification with hydroquinone and aromatic moieties, to enhance binding affinity and stability. Molecular docking studies were conducted using tyrosinase crystal structures (PDB IDs: 3NQ1 and 5I38) to identify key binding interactions. Six lead compounds were synthesized and structurally characterized via NMR and FTIR spectroscopy. Their tyrosinase inhibitory activity was assessed in vitro using mushroom tyrosinase under standardized assay conditions.
Results: Molecular docking revealed favorable interactions within the tyrosinase active site, with compound 9 exhibiting the highest docking scores. In vitro assays confirmed compound 9 as the most potent inhibitor, with an IC₅₀ value of 31.35 µg/ml, outperforming kojic acid (IC₅₀ = 94.96 µg/ml) and hydroquinone (IC₅₀ = 772.8 µg/ml) under identical conditions.
Conclusion: Compound 9 demonstrated superior tyrosinase inhibition compared to conventional agents, indicating its potential as a safer and more effective alternative for treating hyperpigmentation. These findings support further development of compound 9 for use in topical formulations targeting melasma and related pigmentary disorders, with planned follow-up studies including in vivo efficacy and safety evaluations.
Keywords: Tyrosinase inhibitors, Skin whitening, Molecular docking, In vitro assay, Cinnamic acid, depigmentation
© 2026 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2026v18i1.54475 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Hyperpigmentation is a common skin condition characterized by darkened patches due to excess melanin production [1]. It affects people of all skin types and appears in forms like melisma, post-inflammatory hyperpigmentation, and solar lentigines [2-5]. The enzyme tyrosinase, a copper-containing protein in melanocytes, plays a central role in melanogenesis, the process of melanin synthesis by catalyzing the conversion of tyrosine to DOPA and then to dopaquinone [6, 7]. This process is influenced by genetic, hormonal, and environmental factors, with UV radiation being a major trigger [8]. Overproduction of melanin can lead to uneven pigmentation, underscoring the need for safe and effective depigmenting agents [9-11].
Hydroquinone and konjac reduce melanin production primarily by inhibiting tyrosinase, the key enzyme in melanogenesis [12, 13]. Hydroquinone, a phenolic compound, acts as a structural analog of melanin precursors, blocking enzymatic activity and promoting melanosome degradation, with selective cytotoxic effects on melanocytes [14]. Despite its effectiveness, hydroquinone is associated with adverse effects such as skin irritation, post-inflammatory hyperpigmentation, and, in severe cases, exogenous ochronosis [15, 16]. Concerns over its potential carcinogenicity and systemic toxicity have led to its restriction or ban in several countries for over-the-counter use, prompting the search for safer alternatives like konjac [17].
Melanogenesis, the process of melanin production in melanocytes, is tightly regulated by transcriptional, enzymatic, hormonal, and environmental factors. It begins in melanosomes and is primarily driven by tyrosinase, along with TRP-1 and TRP-2. The microphthalmia-associated transcription factor (MITF) plays a central role by regulating melanogenic enzyme expression. MITF activity is modulated by the cAMP/PKA signaling pathway, which is activated when α-MSH binds to the MC1R receptor, leading to increased cAMP levels, PKA activation, and CREB phosphorylation, ultimately enhancing MITF transcription and melanin synthesis [18].
Melanogenesis is also regulated by key signaling pathways. The Wnt/β-catenin pathway promotes melanocyte differentiation and increases MITF expression, a critical transcription factor in melanin synthesis. The MAPK pathway has dual roles—certain MAPKs, like ERK can inhibit melanogenesis by promoting MITF degradation. Additionally, UV radiation stimulates melanogenesis by enhancing α-MSH secretion from keratinocytes and linking pigment production to DNA repair and photoprotection [19].
Hormonal and inflammatory signals also regulate melanogenesis. Estrogens and androgens influence pigmentation through nuclear hormone receptors, while cytokines like TNF-α and IL-1α suppress melanin production. In contrast, endothelin-1 and nitric oxide stimulate melanogenesis via melanocyte signaling pathways. Oxidative stress plays a dual role-low level of ROS can enhance melanin synthesis through stress-activated kinases, whereas excessive ROS can damage melanocytes, leading to hypopigmentation. These multilayered regulatory systems ensure precise control of melanogenesis in response to both internal and external stimuli, maintaining pigmentation balance and adapting to environmental stressors like UV exposure [20].
Hydroquinone and konjac inhibit tyrosinase, the key enzyme in melanin synthesis, thereby reducing pigmentation. Hydroquinone, a phenolic compound, also promotes melanosome degradation and can be selectively toxic to melanocytes. Despite its effectiveness, hydroquinone is linked to adverse effects such as skin irritation, post-inflammatory hyperpigmentation, and, in severe cases, exogenous ochronosis. Concerns over its potential carcinogenicity and systemic toxicity have led to regulatory restrictions or bans in several countries [21].
Konjac, derived from Amorphophallus konjac tubers, contains glucomannan and other bioactive compounds that mildly inhibit tyrosinase. Unlike strong inhibitors like hydroquinone, konjac acts more indirectly, possibly by interfering with oxidative steps or through antioxidant activity that reduces melanin-triggering oxidative stress. It is generally considered safer and less irritating, though mild allergic reactions may occur in sensitive individuals, especially when using concentrated or unrefined forms [22].
Various compounds inhibit tyrosinase through different mechanisms. Glycolic acid acts as a mixed-type inhibitor by altering enzyme conformation, while heterocyclic compounds inhibit based on their bioactive structures [23]. Konjac, though milder, reduces melanin synthesis indirectly via antioxidant effects and is generally safer than hydroquinone [23].
Natural compounds, particularly cinnamic acid and its derivatives, have emerged as promising alternatives due to their wide availability, low toxicity, and diverse biological properties [24, 25].
These derivatives, commonly found in plants, have been explored for their potential applications in both cosmetics and pharmaceuticals. Notably, research has shown that certain cinnamic acid derivatives possess tyrosinase-inhibitory activity, making them strong candidates for depigmenting treatments [26, 27]. By modulating the melanogenic pathway, these inhibitors have the potential to reduce melanin synthesis while mitigating the toxic effects linked to conventional therapies [28, 29]. We hypothesize that esterification of cinnamic acid with hydroquinone-derived moieties will enhance binding affinity while reducing oxidative degradation.
Recent advances in hyperpigmentation treatment focus on dual-target tyrosinase inhibitors and nanoparticle-based delivery systems to improve efficacy, stability, and skin penetration. Compounds like MHY498 and Sepiwhite®, when encapsulated in solid lipid nanoparticles (SLNs), show enhanced tyrosinase inhibition and melanin reduction [30, 31]. Other platforms like liposomes, penetration enhancer vesicles (PEVs) [32], and ethosomal gels improve skin retention and controlled release of agents like alpha-arbutin [33]. Additionally, pH-responsive ZnO quantum dots grafted with BQ-788 enable targeted delivery of ellagic acid, offering promising results in melanin suppression [34]. These strategies collectively offer safer and more effective options for managing hyperpigmentation.
This study investigates the potential of novel cinnamic acid-derived compounds as tyrosinase inhibitors using a multidisciplinary approach that includes computational modeling, chemical synthesis, and in vitro evaluation. Molecular docking was employed to identify candidates with strong interactions at the tyrosinase active site, followed by synthesis and biological testing of the most promising compounds. The goal is to develop safe, effective, and stable depigmenting agents with potential pharmaceutical applications.
MATERIALS AND METHODS
Computational methods
Protein structure preparation
The crystal structures of human tyrosinase (PDB codes 3NQ1 and 5I38) were retrieved from the Protein Data Bank (PDB) [14–16] for computational analysis. These structures were specifically selected based on their high resolutions (3.0 Å for 3NQ1 and 2.7 Å for 5I38), their relevance to human tyrosinase function, and the presence of co-crystallized inhibitors (KOJ1351 and KOJ303), which facilitated accurate identification of the catalytic site and validation of docking protocols.
Before docking, protein preparation was carried out using Discovery Studio 2022 (DassaultSystèmes BIOVIA) [17]. Preparation steps included removal of water molecules, addition of hydrogen atoms, standardization of atom names, and correction of missing loops and incomplete residues. Protonation was performed at pH 7.4 to ensure appropriate charge states, and bond orders were verified. The structural refinement was completed through energy minimization using the CHARMm force field, ensuring that the proteins were in a stable and optimized state for docking analysis.
Ligand preparation
A set of ten novel cinnamic acid-derived compounds was designed as potential tyrosinase inhibitors. The initial 2D structures were drawn using ChemDraw Ultra 7.0 (CambridgeSoft) and subsequently converted into 3D conformations using Discovery Studio 2022. Ligands were further refined using the Prepare Ligand protocol, which accounted for ionization states and tautomeric forms within a pH range of 7.2 to 7.6, thereby generating biologically relevant protonation states suitable for accurate docking simulations.
Molecular docking
Docking studies were initially performed using the LibDock algorithm in Discovery Studio 2022, wherein the co-crystallized ligands were removed from the active sites, and the prepared ligands were docked into the defined binding pockets of both tyrosinase structures. For each compound, ten binding conformations were generated, and the docking scores and interaction profiles (e.g., hydrogen bonding, π-π stacking, and hydrophobic contacts) were analyzed.
To enhance the robustness and validity of the results, a secondary docking analysis was performed using the CDocker algorithm. This method confirmed the binding affinities and provided detailed interaction energies and binding poses for each compound [35, 36].
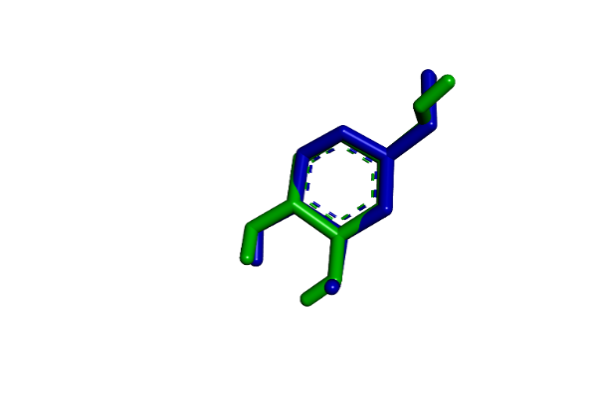
To assess the reliability and reproducibility of the docking protocols, the native co-crystallized ligands (KOJ1351 in 3NQ1 and KOJ303 in 5I38) were re-docked into their respective active sites using AutoDockVina (version 1.2.3). A grid box size of 22 Å × 22 Å × 22 Å was defined to encompass the entire catalytic pocket, and exhaustiveness was set to 8. The RMSD values between the re-docked and crystallographic poses were 1.20 Å for KOJ1351 and 0.97 Å for KOJ303, confirming accurate pose prediction within the accepted threshold of 2.0 Å (fig. 1E, 1F).
All docking results were further visualized and analyzed using Discovery Studio Visualizer 2022 and PyMOL (version 2.5.0) to interpret binding orientations and key residue interactions.
Chemical synthesis
Materials and reagents
The following chemical reagents were obtained from Sigma-Aldrich: Thionyl chloride (SOCl2), dicyclohexylcarbodiimide (DCC) (C13H22N2), 4-dimethylaminopyridine (DMAP) ((CH3)2NC5H4N), mushroom tyrosinase enzyme (CAS Number: 9002102), and L-DOPA substrate (CAS Number: 333786). AZ Chem for Chemicals supplied hydroquinone (C6H6O2), dichloromethane (CH2Cl2), and chloroform (CHCl3). Ethyl acetate (CH3COOCH2CH3) and n-hexane (C6H14) were obtained from Tedia® HPLC/Spectro. Additionally, Lab Chem provided high-performance liquid chromatography (HPLC) grade water and gas chromatography (GC) grade methanol (CH3OH). Kojic acid (C6H6O4), mefenamic acid (C15H15NO2), and cinnamic acid (C9H8O2) were sourced from Genochem World.
General procedure for synthesizing compounds (5, 6, 7, and 8)
The target compounds were synthesized via esterification involving mefenamic acid and/or hydroquinone, utilizing DCC as a coupling reagent and DMAP as a nucleophilic catalyst. Initially, carboxylic acid was dissolved in dichloromethane, followed by the gradual addition of DCC under continuous stirring and cooling. Hydroquinone and/or kojic acid were subsequently introduced into the reaction mixture, which was allowed to stir at room temperature for 18 h. Upon completion, the resulting dicyclohexylurea precipitate was removed via filtration. The organic layer was sequentially washed with 1M hydrochloric acid and neutralized with sodium bicarbonate. The product was extracted, dried over magnesium sulfate, and purified through thin-layer chromatography (TLC). The specific synthetic schemes are illustrated in scheme 5.
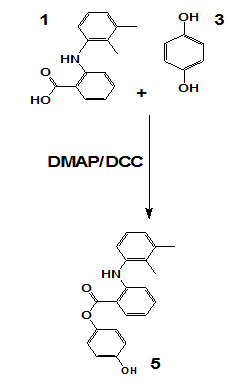
Scheme 1: Synthesis of compound
Scheme 5 represents a general Steglich esterification, where mefenamic acid or hydroquinone derivatives were coupled with cinnamic acid analogs using dicyclohexylcarbodiimide (DCC) as the coupling agent and 4-dimethylaminopyridine (DMAP) as the nucleophilic catalyst. This method was chosen for its mild conditions and compatibility with sensitive functional groups.
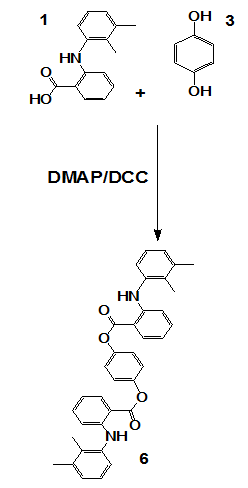
Scheme 2: Synthesis of compound 6
OR
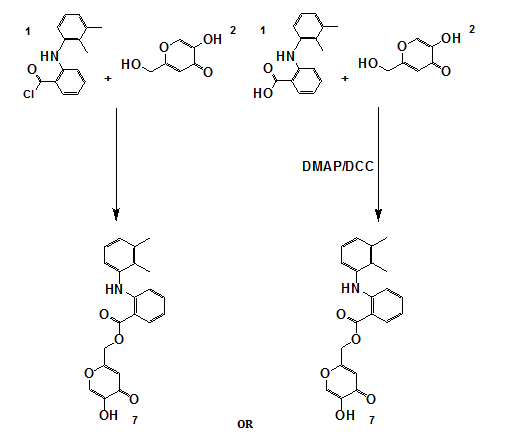
Scheme 3: Synthesis of compound 7
OR
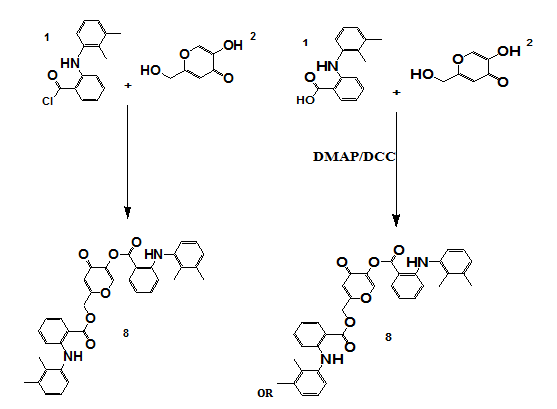
Scheme 4: Synthesis of compound 8
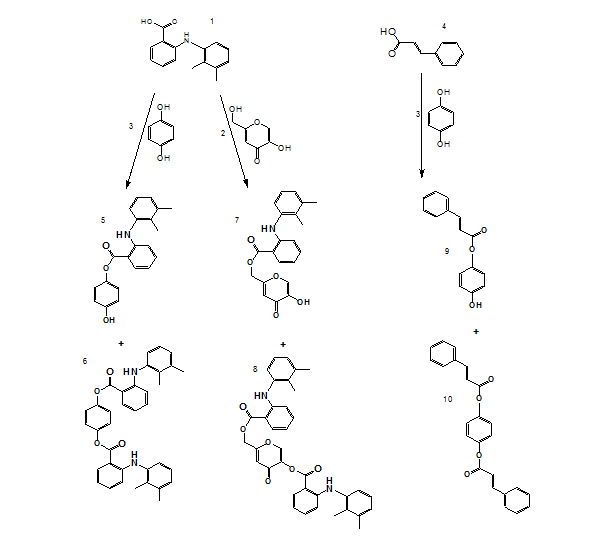
Scheme 5: General scheme for synthesis of compounds (1-10)
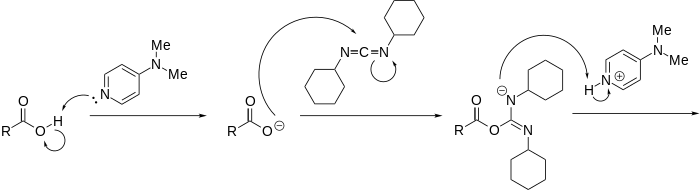
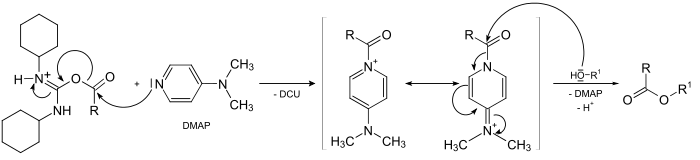
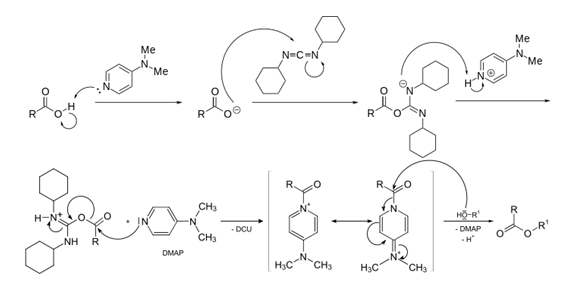
Scheme 6: DMAP and DCC mechanism of action
Synthesis of compounds 9 and 10
Scheme 7 and Scheme 8 detail the synthesis of compounds 9 and 10, respectively. Where Compounds 9 and 10 were synthesized via esterification of hydroquinone with cinnamoyl chloride in the presence of DMAP in an inert nitrogen atmosphere to prevent moisture-induced hydrolysis. The preparation of cinnamoyl chloride involved reacting cinnamic acid with thionyl chloride. For compound 9, hydroquinone was dissolved in dichloromethane, cooled, and cinnamoyl chloride was introduced dropwise in the presence of triethylamine and DMAP. The reaction mixture was stirred for 4 h, followed by purification through filtration, organic extraction, drying with magnesium sulfate, and TLC. The synthesis of compound 10 was carried out by heating cinnamyl chloride and hydroquinone in dichloromethane at 70 °C for 6 h with DMAP as a catalyst. The final product was purified through sequential washing, filtration, drying, monitored using thin-layer chromatography (TLC), and purified by column chromatography. Spectroscopic characterization was performed using NMR (¹H and ¹³C), FTIR, and HRMS, confirming the expected structures. Detailed spectral data are provided in the supplementary section.
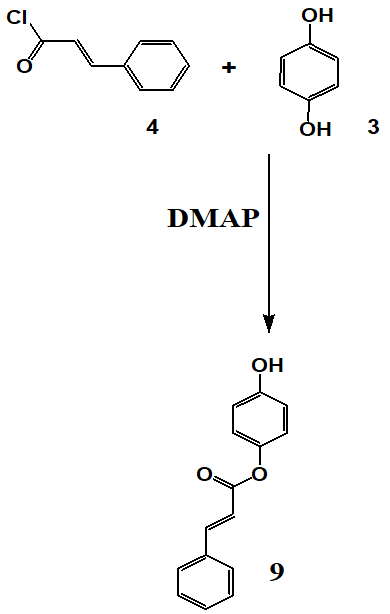
Scheme 7: Synthesis of compound 9
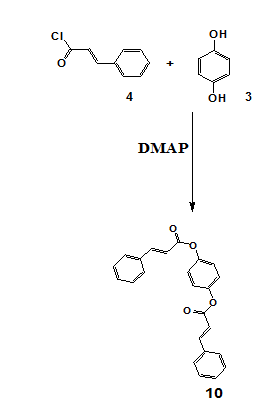
Scheme 8: Synthesis of compound 10
Characterization
The structural and compositional properties of the synthesized compounds were analyzed using various spectroscopic techniques. Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker Avance III 500 spectrometer at the University of Jordan. Fourier-transform infrared (FTIR) spectra were obtained using a Shimadzu 8400F FT-IR spectrophotometer at Isra University, Faculty of Pharmacy (Jordan). High-resolution mass spectrometry (HRMS) data were collected utilizing the Bruker Daltonik Impact II ESI-Q-TOF system.
Specific NMR data for synthesized compounds
Compound 7
IR: 2950 (C-H) aromatic,1732(C=O),1045 (C-O).
1H NMR (500MHz, CDCl3) δ 2.10-2.28 (6H, 2.15-2.32 s),(5.1-5.2 2H, s),(6.89 1H, s), 6.70-7.04 (3H, 6.77 (dd, J = 7.8, 2.2 Hz), 6.83 (dd, J = 7.9, 2.2 Hz), 6.92 (t, J = 7.8 Hz)), 7-7.15 (2H, 7.04 (s), 7.08 (d, J = 8.6, 1.3, 0.5 Hz)), 7.37 (1H, ddd, J = 7.9, 7.5, 1.3 Hz), 7.48-7.71 (2H, 7.54 (ddd, J = 7.9, 1.3, 0.5 Hz), 7.64 (ddd, J = 8.6, 7.5, 1.3 Hz)),7.67-7.7(s,1H), 7.9-8.1(m,1H),8.13-8.14 (s,1H) 8.97-9.1(s,1H).
13C NMR: (125MHZ, CDCl3) δ 14.0 (1C), 20.0 (1C), 60.8 (1C), 110.5 (1C), 111.7 (1C), 117.3-117.3 (2C, 117.3), 117.3)), 128.2 (1C), 128.4 (1C), 128.8-129.1 (2C, 128.9, 129.0), 131.0 (1C), 132.9 (1C), 138.4 (1C), 138.8 (1C), 139.8 (1C), 146.4 (1C), 147.7 (1C),165.4 (1C),168.0 (1C, C=O),173.5 (1C).
HRMS: (ESI, positive mode): m/z(M++H+):Found 366.13(C21H19NO5) Requires 365.0
Compound 8
IR: 2983(C-H) aromatic,1738(C=O), 1057 (C-O).
1H NMR: (500MHZ-CDCL3) δ 2.10-2.32 (12H, 2.15 (s), 2.15 (s), 2.23 (s), 2.32 (s)), 4.37-4.38 (2H, s), 5.23 (1H, s), 6.68-6.74 (6H, 6.68 (dd, J = 7.8, 2.2 Hz), 6.7 (dd, J = 7.8, 2.2 Hz), 6.73 (dd, J = 7.9, 2.2 Hz), 6.74 (dd, J = 7.9, 2.2 Hz), 7.05 – 7.12 (t, J = 7.7 Hz, 2H), 7.12 – 7.15 (m, 2H), 7.15 – 7.26 (m, 2H), 7.26– 7.3 (m, 2H), 8.01 – 8.07 (ddd, J = 1.2, 8.2, 9.8 Hz, 2H), 8.07 – 8.13(s, 1H), 8.13 – 8.15 (s, 1H).
13C NMR(125MHZ-CDCL3) δ 14-14.1 (2C, 14.0), 14.1), 20-20.5 (2C, 20.0), 20.0), 60.8 (1C), 110.5 (1C), 111.7-111.8 (2C, 111.7), 111.7)), 117.3-117.3 (4C, 117.3), 117.3), 117.3), 117.3), 128.1-128.3 (2C, 128.2), 128.2), 128.3-128.5 (2C, 128.4), 128.4), 128.8-129.1 (4C, 128.9), 128.9), 129.0), 129.0), 130.9-131.1 (2C, 131.0), 131.0), 132.8-133.0 (2C, 132.9), 132.9), 138.3-138.5 (2C, 138.4), 138.4 (s)), 138.8-138.9 (2C, 138.8), 138.8), 139.3 (1C), 140.5 (1C), 147.6-147.8 (2C, 147.7), 147.7), 162.4 (1C), 165.0 (1C, C=O), 167.2 (1C,C=O), 172.7 (1C).
HRMS: (ESI, positive mode): m/z(M++H+):Found 589.22 (C36H32N2O6) Requires 588.3
Compound 5
IR: 2930 (C-H) aromatic,1744(C=O),1047 (C-O).
1H NMR: (500MHZ-CDCL3) δ 2.29-2.48(6H, 2.15 (s), 2.48(s)), 6.80-6.86(8H, 6.77 (dd, J = 7.8, 2.2 Hz), 6.83 (dd, J = 7.9, 2.2 Hz), 6.85 (ddd, J = 8.6, 2.7, 0.5 Hz), 6.92 (t, J = 7.8 Hz), 7.02 7.07 (ddd, J = 8.2, 1.3, 0.6 Hz)), 7.37 (1H, ddd, J = 7.9, 7.5, 1.3 Hz), 7.4-7.46(2H, 7.55 (ddd, J = 7.9, 1.3, 0.6 Hz), 7.64 (ddd, J = 8.2, 7.5, 1.3 Hz),7.94-7.99(dt, J = 0.7, 8.1 Hz, 1H), 8.04 – 8.15(s, 1H).
13C NMR: (125MHZ –CDCL3) δ 14.0 (1C), 20.0 (1C), 111.7 (1C), 115.5 (2C), 115.8 (2C), 117.3-117.3 (2C, 117.3), 117.3), 128.2 (1C), 128.4 (1C), 128.8-129.1 (2C, 128.9), 129.0)), 131.0 (1C), 132.9 (1C), 138.4 (1C), 138.8 (1C), 147.7 (1C), 150.6 (1C), 153.4 (1C), 173.2 (1C, C=O).
HRMS: (ESI, positive mode): m/z(M++H+):Found 334.14(C21H19NO3) Requires 333.12
Compound 6
IR: 2959 (C-H) aromatic,1733(C=O),1046 (C-O).
1H NMR: (500MHZ-CDCL3) δ 2.20-2.4 (12H, 2.15 (s), 2.23 (s)), 6.78-6.98 (6H, 6.77 (dd, J = 7.8, 2.2 Hz), 6.83 (dd, J = 7.9, 2.2 Hz), 6.92 (t, J = 7.8 Hz)), 7.00-7.23 (6H, 7.07 (ddd, J = 8.2, 1.3, 0.6 Hz), 7.17 (ddd, J = 8.3, 1.9, 0.5 Hz)), 7.37 (2H, ddd, J = 7.9, 7.5, 1.3 Hz), 7.44-7.65 (4H, 7.55 (ddd, J = 7.9, 1.3, 0.6 Hz), 7.65 (ddd, J = 8.2, 7.5, 1.3 Hz), (dt, J = 0.7, 8.1 Hz, 1H), 8.1–8.17(s, 1H),8.31-8.32(s,1H).
13C NMR: (125MHZ –CDCL3) δ 14.0 (2C), 20.6 (2C), 109.7 (2C), 113.5 (4C), 116.3-117.3 (4C, 116.3), 116.3), 128.2 (2C), 128.4 (2C), 126.8-127.1 (4C, 128.9), 129.0), 131.0 (2C), 132.9 (2C), 138.4 (2C), 138.8 (2C), 148.7 (2C), 150.6 (2C), 167.25 (2C, C=O).
HRMS: (ESI, positive mode): m/z(M++H+):Found 557.4 (C36H32N2O4) Requires 556.3.
Compound 9
IR: 2964 (C-H) aromatic,1737(C=O),1058(C-O).
1H NMR: (500 MHz, CDCL3) δ 6.59 (d, J = 16.1 Hz, 1H), 6.80 – 6.87 (m, 2H), 6.92 – 6.96 (m, 2H), 7.32 – 7.42 (m, 3H), 7.50 – 7.57 (ddt, J = 1.6, 4.6, 6.4 Hz, 2H), 7.81 – 7.87 (m, 1H).
13C NMR: (125 MHz, CDCL3) δ 116.1 (1C), 117.24 (2C), 127.2 (2C), 127.8 (1C), 128.3 (2C,), 130.7 (1C), 134.6 (1C), 144.6 (1C), 146.6 (1C) 153.4 (1C), 166.7 (1C, C=O).
HRMS: (ESI, positive mode):m/z(M++H+):Found 241.08(C15H12O3) Requires 240.0.
Compound 10
IR: 2964.8 (C-H) aromatic,1732.6(C=O),1044.7 (C-O).
1H NMR: (500 MHz, CDCL3) δ 6.55 (2H, d, J = 16.3 Hz), 7.16 (4H, ddd, J = 8.3, 1.9, 0.5 Hz), 7.31-7.55 (10H, 7.38 (ddd, J = 8.0, 2.3, 1.5, 0.5 Hz), 7.64 (dd, J = 8.0, 7.3, 2.0, 0.5 Hz), 7.71 (tt, J = 7.3, 1.5 Hz)), 7.9 (2H, d, J = 16.3 Hz).
13C NMR (125 MHz, CDCL3) δ 117.12 – 118.54, 122.33 – 122.45, 128.72 – 128.84, 129.00 –129.12, 130.00 – 130.12, 134.06 – 134.18, 144.59, 144,89, 165.27 – 167.4(2C, C=O).
HRMS: (ESI, positive mode): m/z(M++H+):Found 388.15 OR 371.12 (C24H18O4)+NH4, H+Requires 370.1.
In vitro tyrosinase assay
Reagents
Mushroom tyrosinase enzyme, L-DOPA, and all other necessary reagents were obtained from Sigma-Aldrich.
Tyrosinase inhibition assay
The inhibitory activity of the synthesized compounds against mushroom tyrosinase was assessed using a modified spectrophotometric method based on the work of Masamoto et al. where the substrate concentration (L-DOPA) was used at 0.9 mg/ml (approximately 4.6 mmol), which is higher than in the original method to ensure sufficient spectrophotometric detection. And the reaction was incubated for 10 min at 27 °C. The assay was conducted in a 96-well microplate, with each well containing 10 μl of the test compound dissolved in either dimethyl sulfoxide (DMSO) or methanol, depending on its solubility and it is included in all experiments to ensure that the solvents did not interfere with enzyme activity. Additionally, 60 μl of phosphate buffer (50 mmol/l, pH 6.8) and 20 μl of L-DOPA solution (0.9 mg/ml in phosphate buffer) were added. The reaction was initiated by introducing 10 μl of mushroom tyrosinase enzyme (1000 U/ml in phosphate buffer) into the mixture, since it’s known that there is limitation of using mushroom tyrosinase, but it is a widely accepted model for initial screening due to its availability and cost-effectiveness. The microplate was incubated at 27 °C for 10 min, after which the formation of dopachrome was quantified spectrophotometrically by measuring absorbance at 450 nm using a FluoStar Omega microplate reader.
Data analysis
The percentage of tyrosinase inhibition for each compound was calculated using the following equation:
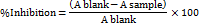
were represents the absorbance in the presence of the inhibitor, while corresponds to the absorbance of the control (without inhibitor)[7]. The half-maximal inhibitory concentration (IC50), defined as the concentration required to achieve 50% inhibition of tyrosinase activity, was determined using GraphPad Prism 8 software.
RESULTS
Molecular docking results
Molecular docking studies were performed to evaluate the binding affinity of the synthesized compounds with tyrosinase protein crystal structures, specifically those with PDB codes 3NQ1 and 5I38. The docking simulations were carried out using AutoDockVina, employing an energy-minimized ligand structure and a grid box centered on the active site of the protein. The docking protocol was validated by re-docking the native ligand, ensuring accurate binding mode prediction.
Binding interactions, including hydrogen bonding, hydrophobic interactions, and π-π stacking, were analyzed using Discovery Studio Visualizer and PyMOL software. The docking scores (binding energies) were recorded to compare the potential inhibitory activity of the synthesized compounds against tyrosinase. Results indicated that certain derivatives exhibited strong interactions with the catalytic residues of tyrosinase, suggesting their potential as effective inhibitors. Further validation through molecular dynamics simulations and free energy calculations may be required to confirm these findings.
Table 1 and 2 summarizes the LibDock and CDocker scores for the selected compounds. Among them, compound 9 exhibited the highest docking scores for both tyrosinase structures, indicating significant binding potential. The docking studies confirmed that the synthesized compounds occupied the active site of tyrosinase, reinforcing their inhibitory activity. Compound 5 also displayed promising binding affinity, although with lower scores than compound 9. Reference compounds 1, 2, 3, and 4 exhibited strong binding interactions, albeit slightly lower than compound 9. Conversely, compounds 6, 7, 8, and 10 showed weaker docking results, with "ND" (not detected) indicating an absence of successful binding for both LibDock and CDocker analyses.
Table 1: Ligand docking scores and interaction energies for tyrosinase (3NQ1) and (5I38), a comprehensive evaluation of reference compounds
| Compound name | Chemical structure | Libdock score (3NQ1) tyrosinase |
CDocker score (3NQ1) tyrosinase |
Libdock score (5I38) Tyrosinase |
CDocker score (5I38) tyrosinase |
| 1 |
|
32.3608±5.026 | 24.8625±1.145 | 75.1984± | 30.4152±2.654 |
| 2 | 53.2076±3.784 | 17.5296±0.879 | 80.0407±3.654 | 26.0066±3.778 | |
| 3 | 39.345±1.345 | 14.616±2.071 | 65.1616±4.510 | 25.7469±6.25 | |
| 4 | 60.1024±5.960 | 19.949±1.037 | 72.2378±10.612 | 30.4931±11.621 |
Table 2: Ligand docking scores and interaction energies for tyrosinase (3NQ1) and (5I38) comprehensive evaluation of synthesized derivatives
| Compound name | Chemical structure | Libdock score (3NQ1) tyrosinase |
CDocker score (3NQ1) tyrosinase |
Libdock score (5I38) tyrosinase | CDocker score (5I38) tyrosinase |
| 7 |  |
ND** | ND | ND | ND |
| 8 | ND | ND | ND | ND | |
| 5 | 58.6618±12.074 | 29.2596±9.123 | ND | 23.8715±2.065 | |
| 6 | ND | ND | ND | ND | |
| 9 | 55.9954±4.445 | 25.6128±0.985 | 80.3017±12.272 | 36.8347±2.774 | |
| 10 | ND | ND | ND | ND |
**Not Successfully Docked (ND)
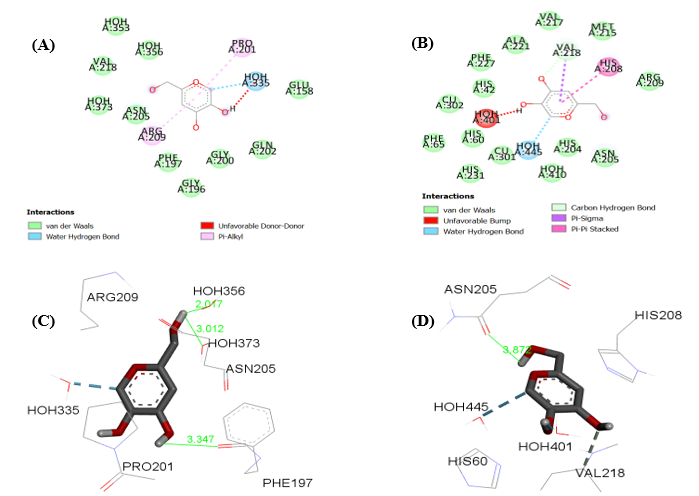
An in-depth examination of ligand–protein interactions has highlighted the pivotal roles of hydrogen bonding, hydrophobic contacts, and π-π stacking in determining binding strength. The interaction profiles of the co-crystallized ligands KOJ303 and KOJ1351 within the active sites of tyrosinase structures 5I38 and 3NQ1, respectively, offer valuable insights into these mechanisms.
In the case of KOJ303 bound to 5I38, the 2D interaction diagram reveals that the ligand is stabilized through several water-mediated hydrogen bonds involving HOH335, HOH356, and HOH373. These interactions help anchor the molecule within the binding cavity. Additionally, π-alkyl interactions with residues such as PHE197 and GLY200 contribute to the hydrophobic environment that supports ligand retention. Although a donor–donor clash is observed, likely due to spatial proximity between donor atoms, it appears to exert minimal influence on the overall binding efficacy.
For KOJ1351 in the 3NQ1 complex, the interaction map shows a network of hydrogen bonds with water molecules, particularly HOH445 and HOH401. The ligand also engages in π–π stacking and π–sigma interactions with aromatic residues like HIS208 and VAL218, which enhance its fit within the hydrophobic core. A slight steric overlap with HOH401 is noted, but it does not significantly compromise the ligand’s affinity.
The three-dimensional binding pose of KOJ303 in 5I38 further confirms its stable orientation. The ligand forms multiple hydrogen bonds with key residues such as ASN205 and PHE197, as well as with several water molecules. These interactions, with distances ranging from approximately 2.01 to 3.35 Å, align well with the electron density, supporting the accuracy of the crystallographic model.
Similarly, KOJ1351 adopts a conformation in 3NQ1 that complements the active site geometry. It forms hydrogen bonds with ASN205, HOH401, and HOH445, and interacts with HIS208 through π-stacking. The hydrogen bond to ASN205, measured at 3.87 Å, suggests a stable but not overly tight interaction.
Redocking experiments validate the reliability of the docking protocol. For KOJ303 in 5I38, the re-docked pose closely matches the experimental structure, with an RMSD of 0.97 Å. KOJ1351, when redocked into 3NQ1, shows a similarly strong alignment, with an RMSD of 1.20 Å—well within the acceptable range—demonstrating the robustness of the computational approach.
Turning to compound 9, its binding profile within the tyrosinase active site is particularly noteworthy. It forms a conventional hydrogen bond with VAL217 and VAL218 via its hydroxyl group, while π-alkyl interactions with PRO201 and PHE197 stabilize its aromatic moiety. A water-mediated hydrogen bond with HOH373 and several van der Waals contacts, including those with ARG209 and GLU158, further reinforce its binding. These interactions explain its high docking score and potent inhibitory activity.
Compound 5 also exhibits a favorable binding mode, characterized by π–π stacking with PHE197 and hydrogen bonding with HOH373. Additional hydrophobic interactions with MET184 and GLY200 support its moderate activity, despite a slightly lower docking score than compound 9.
In contrast, compound 3 shows a more limited interaction profile. It forms a single hydrogen bond with GLY216 and a π-cation interaction with ARG209. While the latter suggests some electrostatic stabilization, the absence of extensive hydrogen bonding or hydrophobic contacts correlates with its weaker binding and lower biological activity.
Compound 2, though forming two strong hydrogen bonds with GLY216 and HOH335 and engaging in π-alkyl interactions with ARG209 and ASN205, lacks the structural complexity needed for broader interaction. This limits its potency relative to compound 9.
 (E)
(E)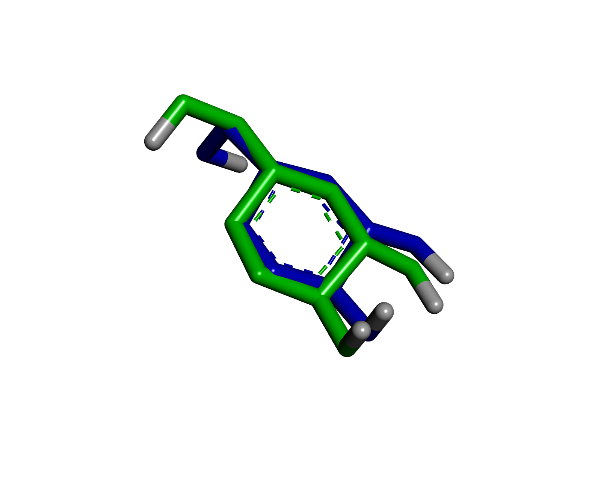 (F)
(F)
Fig. 1: (A) 2D-analysis of cocrystallized ligand (KOJ303) into the binding pocket (5I38), (B) 2D-analysis of cocrystallized ligand (KOJ1351) into the binding pocket (3NQ1), (C) 3D-analysis of cocrystallized ligand (KOJ303) into the binding pocket (5I38), (D) 3D-analysis of cocrystallized ligand (KOJ1351) into the binding pocket (3NQ1). (E) redocking results of co-crystallized ligand (KOJ303) (green) with the docked pose (blue) into the binding site of 5I38 (RMSD = 0.97Å) (F) redocking results of co-crystallized ligand (KOJ1351) (green) with the docked pose (blue) into the binding site of 3NQ1 (RMSD = 1.20Å)
The structure–activity relationship (SAR) becomes evident when comparing these compounds. Compound 9, with its rich network of hydrogen bonds and hydrophobic interactions, demonstrates the strongest binding. Compound 5 follows with moderate activity, while compounds 2 and 3, with simpler interaction profiles, show reduced efficacy.
This trend is further supported by docking results using LibDock, where compound 9 again shows the most extensive interaction network in the 3NQ1 structure. The consistency of these findings across different docking tools and protein targets underscores the predictive power of the modeling strategy.
Fig. 4 and 5, which depict docking into 5I38 using CDocker and LibDock, respectively, reinforce these conclusions. Compound 9 consistently forms multiple stabilizing interactions with residues such as ASN205, HOH356, VAL217, VAL218, ARG209, and PHE197, along with aromatic stabilization via PRO201. Compound 5 maintains moderate binding through π-alkyl and water-mediated contacts, while compounds 2 and 3 show limited engagement with key residues, aligning with their lower in vitro activity.
Altogether, the reproducibility of these interaction patterns across docking platforms and protein structures highlights the reliability of the computational approach. Compound 9’s consistent performance positions it as the most promising candidate for tyrosinase inhibition among the series studied.
Chemical synthesis and characterization
The targeted compounds (5, 6, 7, 8, 9, and 10) were successfully synthesized through esterification reactions using DCC as a coupling agent and DMAP as a catalyst. The reaction conditions were optimized to maximize product yields, and thin-layer chromatography (TLC) was used to monitor reaction progress and ensure product purity. Yields varied based on the molecular structure of each compound. The percentage yields for the synthesized compounds ranged from 65% to 88%, depending on the steric and electronic nature of the substituents. Purity of the final compounds was confirmed by HPLC analysis, with all compounds showing a purity of ≥95%.
Characterization of the synthesized compounds was performed using nuclear magnetic resonance (NMR), Fourier-transform infrared (FTIR) spectroscopy, and high-resolution mass spectrometry (HRMS). NMR spectroscopy confirmed the chemical identity of all synthesized compounds. For example, the NMR spectrum of compound 9 revealed characteristic aromatic proton signals and peaks indicative of the presence of a double bond. Additionally, NMR data confirmed the presence of various carbon environments, including the carbonyl carbon (C=O). FTIR spectroscopy further validated the presence of essential functional groups, with a distinct absorption band observed in the 1600–1800 cm⁻¹ region, corresponding to carbonyl groups. HRMS analysis confirmed the molecular weights of all synthesized compounds, aligning with theoretical values.
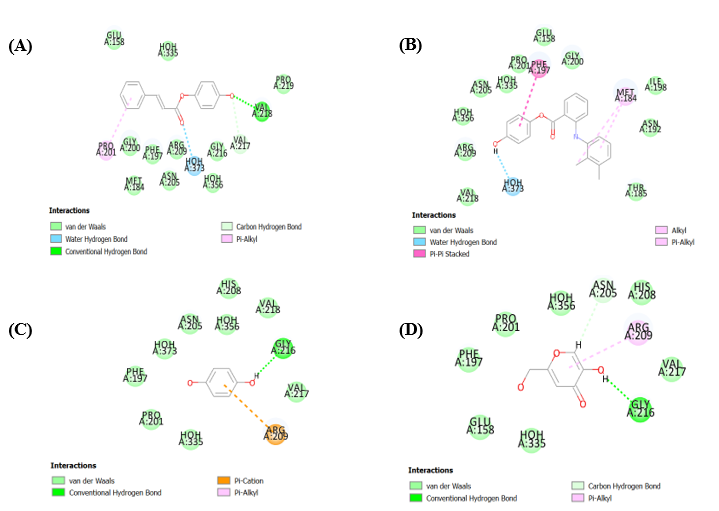
Fig. 2: (A) Docked pose of compound 9 into the binding pocket (3NQ1), (B) Docked pose of compound 5 into the binding pocket (3NQ1), (C) Docked pose of compound 3 into the binding pocket (3NQ1), (D) Docked pose of compound 2 into the binding pocket (3NQ1) using CDocker tool
In vitro tyrosinase inhibition assay
The in vitro inhibitory activity of the synthesized compounds against mushroom tyrosinase was assessed to evaluate their potential as tyrosinase inhibitors[37]. The half-maximal inhibitory concentration (IC50) values were determined to quantify their inhibitory effects. Table 2 presents the IC50 values for compounds 5, 6, 7, and 8, alongside hydroquinone and kojic acid as reference inhibitors. Compound 8 exhibited the most potent tyrosinase inhibition with an IC50 value of 67.9 µg/ml, followed by compound 7 (IC50 = 78.32 µg/ml). Compounds 5 and 6 also demonstrated considerable inhibitory activity, suggesting their potential as effective tyrosinase inhibitors. These results highlight the relevance of the synthesized compounds in the development of novel skin depigmentation agents or therapeutic candidates targeting tyrosinase-associated disorders.
Table 3 also presents the IC50 values of compounds 9, 10, and hydroquinone (HQ). Compound 9 demonstrated a significant inhibitory effect, with an IC50 value of 31.35 µg/ml. In contrast, compound 10 exhibited weak inhibition, with an IC50 value of>500 µg/ml. Hydroquinone, used as a reference compound, displayed an IC50 of 772.8 µg/m. The inhibition profiles of compound 9 and hydroquinone are depicted in fig. 6 and 7. The results indicate that compound 9 exhibited superior tyrosinase inhibition compared to both kojic acid and hydroquinone, highlighting its potential as a potent tyrosinase inhibitor.
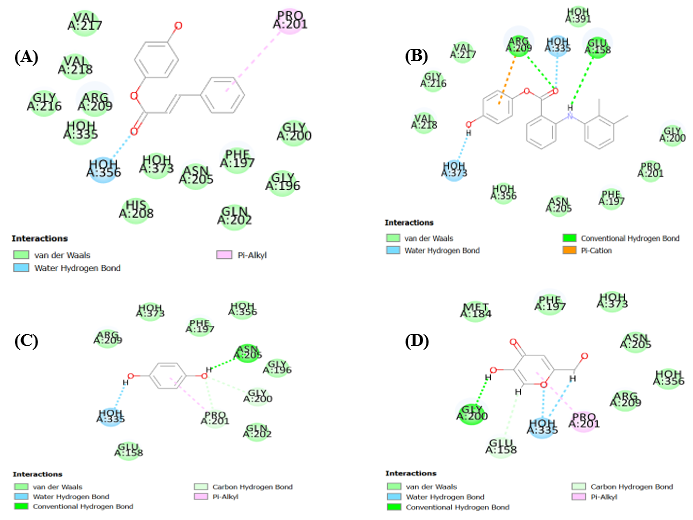
Fig. 3: (A) Docked pose of compound 9 into the binding pocket (3NQ1), (B)Docked pose of compound 5 into the binding pocket (3NQ1), (C) Docked pose of compound 3 into the binding pocket (3NQ1), (D) Docked pose of compound 2 into the binding pocket (3NQ1) using LibDock tool
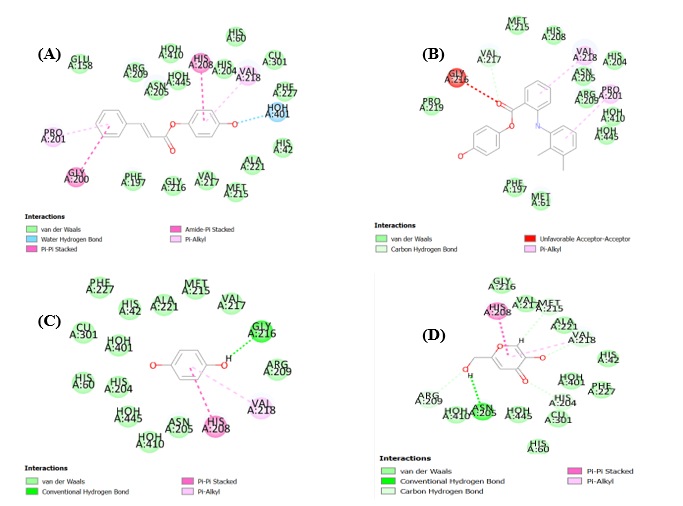
Fig. 4: (A) Docked pose of compound 9 into the binding pocket (5I38), (B) Docked pose of compound 5 into the binding pocket (5I38), (C) Docked pose of compound 3 into the binding pocket (5I38), (D) Docked pose of compound 2 into the binding pocket (5I38) using CDocker tool
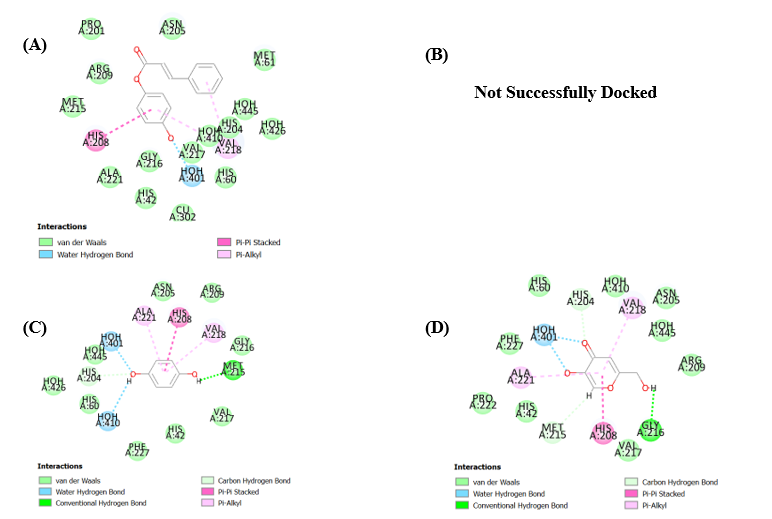
Fig. 5: (A) Docked pose of compound 9 into the binding pocket (5I38), (B) Docked pose of compound 5 into the binding pocket (5I38), (C) Docked pose of compound 3 into the binding pocket (5I38), (D) Docked pose of compound 2 into the binding pocket (5I38) using LibDock tool
Table 3: % Inhibition of tyrosinase enzyme by the prepared compounds 5, 6, 7, 8, 9, and 10
| KojicAcid | ||||||
| Concentration [µg/ml] | 15.625 | 31.25 | 62.5 | 125 | 250 | |
| % Inhibition | 27.30496 | 33.92435 | 41.96217 | 54.37352 | 67.61229 | |
| 22.45863 | 33.80615 | 42.08038 | 50.70922 | 76.59574 | ||
| 24.11348 | 35.10638 | 42.9078 | 57.32861 | 76.24113 | ||
| Average | 24.62569 | 34.27896 | 42.31678 | 54.13712 | 73.48306 | |
| HQ | ||||||
| Concentration [µg/ml] | 25 | 50 | 100 | 200 | 400 | |
| % Inhibition | 1.856764 | 11.19363 | 22.22812 | 39.62865 | 50.66313 | |
| 1.856764 | 10.66313 | 21.90981 | 36.65782 | 56.71088 | ||
| 1.007958 | 11.19363 | 23.28912 | 35.38462 | 53.10345 | ||
| Average | 1.573828 | 11.0168 | 22.47569 | 37.2237 | 53.49248 | |
| 8 | ||||||
| Concentration [µg/ml] | 15.625 | 31.25 | 62.5 | 125 | 250 | |
| % Inhibition | 15.48967 | 34.86635 | 48.66250 | 61.24664 | 80.27314 | |
| 15.02276 | 30.7809 | 48.87358 | 65.68227 | 83.19132 | ||
| 13.27186 | 31.94817 | 46.30559 | 64.74845 | 84.94222 | ||
| Average | 14.59476 | 32.53181 | 47.58959 | 63.89246 | 82.80223 | |
| 7 | ||||||
| Concentration [µg/ml] | 15.625 | 31.25 | 62.5 | 125 | 250 | |
| % Inhibition | 20.04202 | 36.15034 | 40.7027 | 61.01319 | 61.36337 | |
| 22.61002 | 32.41508 | 51.67503 | 55.52702 | 64.28155 | ||
| 21.85012 | 32.88199 | 43.50414 | 61.59683 | 63.11428 | ||
| Average | 21.32602 | 33.8158 | 45.29396 | 59.37901 | 62.91973 | |
| 6 | ||||||
| Concentration [µg/ml] | 15.625 | 31.25 | 62.5 | 125 | 250 | |
| % Inhibition | 9.55414 | 18.47134 | 26.89858 | 39.83341 | 50.0245 | |
| 8.046546 | 19.45125 | 27.97648 | 35.61979 | 50.12249 | ||
| 7.104361 | 20.43116 | 28.66242 | 37.18765 | 52.86624 | ||
| Average | 8.2195 | 19.45125 | 27.84583 | 37.54695 | 51.00441 | |
| 5 | ||||||
| Concentration [µg/ml] | 15.625 | 31.25 | 62.5 | 125 | 250 | |
| % Inhibition | 14.94366 | 22.39098 | 31.79814 | 47.7707 | 55.80598 | |
| 18.56933 | 25.23273 | 30.52425 | 51.88633 | 65.21313 | ||
| 19.05928 | 24.93876 | 31.50416 | 47.18275 | 59.52964 | ||
| Average | 17.52409 | 24.18749 | 31.27552 | 48.94659 | 60.18292 | |
| 9 | ||||||
| Concentration [µg/ml] | 3.90625 | 7.8125 | 31.25 | 62.5 | 125 | |
| % Inhibition | 19.03226 | 28.06452 | 44.08602 | 58.49462 | 68.27957 | |
| 18.60215 | 29.03226 | 48.60215 | 61.39785 | 81.07527 | ||
| 16.98925 | 29.24731 | 49.89247 | 66.77419 | 69.35484 | ||
| Average | 18.20789 | 28.78136 | 47.52688 | 62.22222 | 72.90323 | |
RSD<5%, Number of trials = 3
Table 3: (continued): % inhibition of tyrosinase enzyme by compounds (5,6,7,8,9,10)
| 10 | |||||
| Concentration [µg/ml] | 500 | ||||
| % Inhibition | 34.64373 | ||||
| 37.46929 | |||||
| 36.73219 | |||||
| Average | 36.28174 | ||||
| HQinmethanol | |||||
| Concentration [µg/ml] | 200 | 250 | 400 | 600 | 800 |
| % Inhibition | 3.239587 | 14.13314 | 26.84229 | 36.77465 | 51.83339 |
| 3.773585 | 12.42435 | 26.0947 | 39.12424 | 51.94019 | |
| 4.093984 | 12.95835 | 24.4927 | 36.98825 | 52.15379 | |
| Average | 3.702385 | 13.17195 | 25.8099 | 37.62905 | 51.97579 |
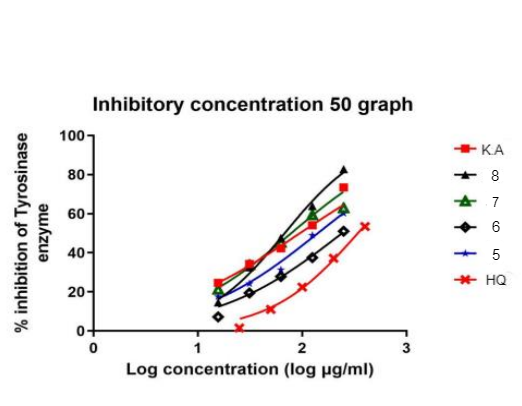
Fig. 6: The tyrosinase inhibition activity IC50 of synthesized compound 5,6,7,8,HQ,K. A
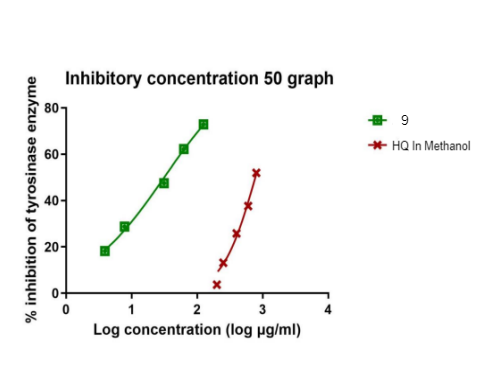
Fig. 7: The tyrosinase inhibition activity IC50 of synthesized compound 9, HQ in methanol
DISCUSSION
This study employs a comprehensive approach to identifying novel tyrosinase inhibitors through computational, synthetic, and biological evaluation. The target compounds were designed and synthesized to enhance the activity of known inhibitors, such as kojic acid, while leveraging the benefits of cinnamic acid derivatives. In silico docking studies provided critical insights into the binding interactions between the synthesized compounds and tyrosinase. The docking analysis confirmed that hydrogen bonding, hydrophobic interactions, and π-π stacking contribute significantly to enzyme stabilization. Among the synthesized derivatives, compound 9 exhibited the strongest binding affinity and docking scores, surpassing the known inhibitor kojic acid. Notably, the cinnamoyl moiety in compound 9 enabled strong π–π stacking interactions with the catalytic residue, which is a key stabilizing force within the active site. This interaction, along with favorable hydrogen bonding and hydrophobic contacts, likely explains the superior docking scores and in vitro activity of compound 9. In contrast, analogs lacking this group or bearing sterically hindered substitutions (e. g., compounds 6–8 and 10) demonstrated weaker activity or failed to dock effectively, supporting a clear structure–activity relationship (SAR) that highlights the importance of planar aromatic systems and ester linkages for optimal binding. The observed SAR, where planar aromatic systems and ester linkages enhance binding, is consistent with findings from Kim et al. (2005), who emphasized the role of π–π stacking and hydrogen bonding in tyrosinase inhibition [15].
The synthesis of the target compounds was carried out efficiently, and their structural integrity and purity were confirmed using TLC and various spectroscopic techniques. The in vitro evaluation of tyrosinase inhibition further validated the computational predictions, with compound 9 demonstrating the most potent inhibitory activity (IC₅₀ = 31.35 µg/ml). This strong correlation between computational and experimental results underscores the reliability of the employed methodologies. While cinnamic acid derivatives have been previously explored Gunia‐Krzyżak et al., 2018 [24], the strategic esterification with hydroquinone in compound 9 represents a novel approach that significantly improves binding affinity and inhibitory potency. This indicate that cinnamic acid derivatives, particularly compound 9, hold significant potential as depigmenting agents. While other synthesized compounds displayed varying degrees of inhibition, compound 9 emerged as the most promising candidate. Additionally, the weaker performance of compounds with lower docking scores aligns with their reduced biological activity, reinforcing the predictive power of molecular docking. The IC₅₀ values of kojic acid (94.96 µg/ml) and hydroquinone (772.8 µg/ml) were determined under identical assay conditions, allowing direct comparison with compound 9. These values further emphasize compound 9’s superior inhibitory potential. Compared to MHY498, which demonstrated significant melanin reduction without cytotoxicity, compound 9 showed comparable IC₅₀ values and superior docking scores, suggesting its potential as a dual-action inhibitor when formulated appropriately [30, 31, 33].
However, the relatively high IC₅₀ of hydroquinone compared to its known clinical efficacy likely reflects limitations of the in vitro model. In vivo, hydroquinone may undergo metabolic activation or redox cycling, forming more reactive species that enhance its activity. This pharmacodynamic behaviour cannot be captured in a simple enzyme assay and should be acknowledged when interpreting these results. It is important to note that mushroom tyrosinase, though widely used for initial screening, differs structurally and functionally from human tyrosinase. This represents a translational limitation, and future studies should include human melanocyte assays or 3D skin models to better predict clinical relevance. Moreover, cytotoxicity profiling was not conducted in the current work. Including assays such as MTT or resazurin-based viability assays on HaCaT keratinocytes or melanocytes would be valuable to assess the safety of these compounds for topical application. Variable interactions observed using different docking tools and protein structures suggest flexibility in binding modes, as well as possible steric limitations that affect activity. Compound 9 showed the highest docking scores using LibDock and CDocker on both proteins (3NQ1 and 5I38): 55.9954, 25.6128, 80.3017, and 36.8347, respectively, higher than kojic acid under identical docking conditions. These results, supported by IC₅₀ values, reinforce the role of in silico docking as a valuable screening strategy before synthesis. Pharmacophore analysis based on the top-scoring pose further suggests that hydrogen bond donors, aromatic rings, and hydrophobic moieties are essential features for potent inhibition. The well-documented inhibitory effects of kojic acid validate the binding interactions observed with its derivative, compound 8. The relatively poor inhibition by hydroquinone in vitro is consistent with its limited solubility and complex behaviour under assay conditions. It's known that cytotoxicity and melanogenic pathway modulation may also contribute to inconsistent outcomes in non-cellular systems [38-42]. Compound 10 displayed markedly reduced tyrosinase inhibition (IC₅₀>500 µg/ml). This poor activity may be attributed to steric hindrance introduced by bulky substituents at the ester linkage, which likely interferes with optimal positioning within the active site. Furthermore, compound 10 exhibited limited solubility in aqueous media, which could compromise bioavailability and effective enzyme binding. These findings suggest that further optimization of substituent size and polarity may enhance activity in future analogs. Given the promising in vitro activity of compound 9, its incorporation into solid lipid nanoparticles or ethosomal gels, as demonstrated with Sepiwhite® and alpha-arbutin, could further enhance its skin permeation and therapeutic efficacy. Overall, these findings highlight the translational potential of compound 9 as a promising lead candidate. Further work should include in vivo efficacy, mechanism-of-action studies, cytotoxicity assessment, and structural optimization to improve selectivity, potency, and safety for hyperpigmentation treatment.
CONCLUSION
This study demonstrated the successful design, synthesis, and evaluation of structurally novel cinnamic acid derivatives as tyrosinase inhibitors to develop safer and more effective treatments for hyperpigmentation. Strategic modifications to the cinnamic acid scaffold, such as esterification with hydroquinone and aromatic moieties, led to enhanced binding affinity and enzyme inhibition. Among the synthesized compounds, compound 9 exhibited the most potent tyrosinase inhibition (IC₅₀ = 31.35 µg/ml), significantly outperforming kojic acid and hydroquinone under identical assay conditions. These findings underscore the potential of compound 9 as a lead candidate for the development of topical formulations targeting melasma and other pigmentary disorders, offering improved efficacy and stability over existing agents.
As a concrete next step, in vivo evaluation using murine models of hyperpigmentation is warranted to validate their efficacy and safety in a biological system. Also, MTT assays on HaCaT keratinocytes and human melanocytes can be conducted to assess cell viability and ensure that the compounds are not cytotoxic at effective concentrations.
ACKNOWLEDGMENT
The authors gratefully acknowledge the financial support provided by Isra University for this research through the Deans Council decision No. (12-23/2022/2023). The research team also extend their gratitude to the Jordan University team for their assistance with NMR analysis and compound characterization. Additionally, they appreciate the support of Nawa Company for their valuable contributions to the in vitro experiments. Also many thanks go to the pharmacy and science faculties at Isra University and Zarqa University for providing access to their research facilities. They also appreciate the Research and Development Department at Zarqa University for supporting the BIOVIA 2022 license, which was instrumental in conducting this study.
The authors are also grateful to the Middle East University, Amman, Jordan, for the financial support granted to cover the publication fee of this article.
AUTHORS CONTRIBUTIONS
For transparency, the individual contributions of all authors are as follows:
Amani Salman is the master’s student from Isra University who conducted the experimental work.
Prof. Manal Najdawi is the main supervisor who provided the research idea, guided every step of the thesis, and wrote the full thesis document.
Dr. Mahmood Al-Sha’er served as co-supervisor, contributed to the modeling studies, data analysis, and co-authored the related sections.
Dr. Rawand Daghmash and Dr. Nawzat Al Jbour were involved in manuscript writing, critical revision, and editing.
Dr. Suliman Edeas and Dr. Anwar Hussein assisted in drafting the first version of the manuscript.
Dr. Zead Abudayeh, Dr. Ala’ Sarhan, and Dr. Loay Hassouneh participated in manuscript review, revisions, and language polishing.
CONFLICT OF INTERESTS
It is to declare that there is no conflict of interest.
REFERENCES
Zaid Alkilani AZ, Musleh B, Hamed R, Swellmeen L, Basheer HA. Preparation and characterization of patch loaded with clarithromycin nanovesicles for transdermal drug delivery. J Funct Biomater. 2023;14(2):57. doi: 10.3390/jfb14020057, PMID 36826856.
Ainger SA, Jagirdar K, Lee KJ, Soyer HP, Sturm RA. Skin pigmentation genetics for the clinic. Dermatology. 2017;233(1):1-15. doi: 10.1159/000468538, PMID 28463841.
Bergfelt DR. Anatomy and physiology of the mare. In: Equine breeding management and artificial insemination. Amsterdam: Elsevier; 2009. p. 113-31. doi: 10.1016/B978-1-4160-5234-0.00011-8.
Prakash V, Jaiswal N, Srivastava M. A review on medicinal properties of Centella asiatica. Asian J Pharm Clin Res. 2017;10(10):69-74. doi: 10.22159/ajpcr.2017.v10i10.20760.
Saadh MJ, Ramirez Coronel AA, Saini RS, Arias Gonzales JL, Amin AH, Gavilan JC. Advances in mesenchymal stem/stromal cell-based therapy and their extracellular vesicles for skin wound healing. Hum Cell. 2023;36(4):1253-64. doi: 10.1007/s13577-023-00904-8, PMID 37067766.
Betz G, Aeppli A, Menshutina N, Leuenberger H. In vivo comparison of various liposome formulations for cosmetic application. Int J Pharm. 2005;296(1-2):44-54. doi: 10.1016/j.ijpharm.2005.02.032, PMID 15885454.
Yousef H, Alhajj M, Sharma S. Anatomy skin (integument) epidermis. StatPearls; 2017. PMID: 29262154.
Chakraborty AK, Funasaka Y, Komoto M, Ichihashi M. Effect of arbutin on melanogenic proteins in human melanocytes. Pigment Cell Res. 1998;11(4):206-12. doi: 10.1111/j.1600-0749.1998.tb00731.x, PMID 9711535.
La Berge GS, Duvall E, Grasmick Z, Haedicke K, Galan A, Leverett J. Recent advances in studies of skin color and skin cancer. Yale J Biol Med. 2020;93(1):69-80. PMID 32226338.
Sauriasari R, Azizah N, Basah K. Tyrosinase inhibition, 2,2-diphenyl-1-picrylhydrazyl radical scavenging activity and phytochemical screening of fractions and ethanol extract from leaves and stem bark of matoa (Pometia pinnata). Asian J Pharm Clin Res. 2017;10(17):85-9. doi: 10.22159/ajpcr.2017.v10s5.23105.
AlJbour ND. An overview of new trends in the cosmetics industry. Int J Appl Pharm. 2025;17(3):136-47. doi: 10.22159/ijap.2025v17i3.53320.
Chang TS. An updated review of tyrosinase inhibitors. Int J Mol Sci. 2009;10(6):2440-75. doi: 10.3390/ijms10062440, PMID 19582213.
Singh K, Bhushan B, Mittal N, Kushwaha A, Raikwar CK, Sharma AK. Recent advances in enzyme inhibition: a pharmacological review. Curr Enzyme Inhib. 2024;20(1):2-19. doi: 10.2174/0115734080271639231030093152.
Kim HD, Choi H, Abekura F, Park JY, Yang WS, Yang SH. Naturally-occurring tyrosinase inhibitors classified by enzyme kinetics and copper chelation. Int J Mol Sci. 2023;24(9):8226. doi: 10.3390/ijms24098226, PMID 37175965.
Kim YJ, Uyama H. Tyrosinase inhibitors from natural and synthetic sources: structure inhibition mechanism and perspective for the future. Cell Mol Life Sci. 2005;62(15):1707-23. doi: 10.1007/s00018-005-5054-y, PMID 15968468.
Melfi F, Carradori S, Angeli A, D Agostino I. Nature as a source and inspiration for human monoamine oxidase B (hMAO-B) inhibition: a review of the recent advances in chemical modification of natural compounds. Expert Opin Drug Discov. 2023;18(8):851-79. doi: 10.1080/17460441.2023.2226860, PMID 37332199.
Hajizadeh M, Moosavi Movahedi Z, Sheibani N, Moosavi Movahedi AA. An outlook on suicide enzyme inhibition and drug design. J Iran Chem Soc. 2022;19(5):1575-92. doi: 10.1007/s13738-021-02416-4.
Sanchez Mas J, Hahmann C, Gerritsen I, Garcia Borron JC, Jimenez Cervantes C. Agonist-independent high constitutive activity of the human melanocortin 1 receptor. Pigment Cell Res. 2004;17(4):386-95. doi: 10.1111/j.1600-0749.2004.00160.x, PMID 15250941.
Gajos Michniewicz A, Czyz M. WNT signaling in melanoma. Int J Mol Sci. 2020;21(14):4852. doi: 10.3390/ijms21144852, PMID 32659938.
Liu T, Xi T, Dong X, Xu D. Research progress on pathogenesis of skin pigmentation in chronic liver disease. Biomol Biomed. 2025;25(6):1218-32. doi: 10.17305/bb.2024.11085, PMID 39689154.
Nordlund JJ, Grimes PE, Ortonne JP. The safety of hydroquinone. J Eur Acad Dermatol Venereol. 2006;20(7):781-7. doi: 10.1111/j.1468-3083.2006.01670.x, PMID 16898897.
Kapoor DU, Sharma H, Maheshwari R, Pareek A, Gaur M, Prajapati BG. Konjac glucomannan: a comprehensive review of its extraction, health benefits and pharmaceutical applications. Carbohydr Polym. 2024;339:122266. doi: 10.1016/j.carbpol.2024.122266, PMID 38823930.
Rubab L. Targeting tyrosinase: heterocyclic compounds in the spotlight. In: Heterocyclic Chemistry-New Perspectives. 2024. p. 45. doi: 10.5772/intechopen.1004439.
Gunia Krzyżak A, Sloczynska K, Popiol J, Koczurkiewicz P, Marona H, Pekala E. Cinnamic acid derivatives in cosmetics: current use and future prospects. Int J Cosmet Sci. 2018;40(4):356-66. doi: 10.1111/ics.12471, PMID 29870052.
Hassan M, Shahzadi S, Kloczkowski A. Tyrosinase inhibitors naturally present in plants and synthetic modifications of these natural products as anti-melanogenic agents: a review. Molecules. 2023;28(1):378. doi: 10.3390/molecules28010378, PMID 36615571.
Juliano CC. Spreading of dangerous skin-lightening products as a result of colourism: a review. Appl Sci. 2022;12(6):3177. doi: 10.3390/app12063177.
Kamboj S, Jhawat V, Saini V, Bala S. Recent advances in permeation enhancement techniques for transdermal drug delivery systems: a review. Curr Drug Ther. 2014;8(3):181-8. doi: 10.2174/15748855113086660012.
Karkeszova K, Mastihubova M, Mastihuba V. Regioselective enzymatic synthesis of kojic acid monoesters. Catalysts. 2021;11(12):1430. doi: 10.3390/catal11121430.
Elliott D. ACCCN’s critical care nursing. 2nd ed. Elsevier Australia; 2011.
Al Amin M, Cao J, Naeem M, Banna H, Kim MS, Jung Y. Increased therapeutic efficacy of a newly synthesized tyrosinase inhibitor by solid lipid nanoparticles in the topical treatment of hyperpigmentation. Drug Des Devel Ther. 2016;10:3947-57. doi: 10.2147/DDDT.S123759, PMID 27980392.
Kabiri H, Tayarani Najaran Z, Rahmanian Devin P, Vaziri MS, Nasirizadeh S, Golmohammadzadeh S. Preparation, characterization and evaluation of anti-tyrosinase activity of solid lipid nanoparticles containing Undecylenoyl phenylalanine (Sepiwhite®). J Cosmet Dermatol. 2022;21(11):6061-71. doi: 10.1111/jocd.15102, PMID 35593521.
Hatem S, Kamel AO, Elkheshen SA, Nasr M, Moftah NH, Ragai MH. Nano-vesicular systems for melanocytes targeting and melasma treatment: in vitro characterization ex-vivo skin retention and preliminary clinical appraisal. Int J Pharm. 2024;665:124731. doi: 10.1016/j.ijpharm.2024.124731, PMID 39306205.
Khan HM. Encapsulation of alpha arbutin a depigmenting agent in nanosized ethosomes: in vitro and in vivo human studies. Heliyon. 2023 Aug 23;9(9):e19326. doi: 10.1016/j.heliyon.2023.e19326.
Huang X, Chen C, Zhu X, Zheng X, Li S, Gong X. Transdermal BQ-788/EA@ZnO quantum dots as targeting and smart tyrosinase inhibitors in melanocytes. Mater Sci Eng C Mater Biol Appl. 2019;102:45-52. doi: 10.1016/j.msec.2019.04.042, PMID 31147016.
Jeyranpour F, Alahyarizadeh G, Arab B. Comparative investigation of thermal and mechanical properties of cross-linked epoxy polymers with different curing agents by molecular dynamics simulation. J Mol Graph Model. 2015;62:157-64. doi: 10.1016/j.jmgm.2015.09.012, PMID 26432014.
Abdullah AH, Alarareh AK, Al Sha ER MA, Habashneh AY, Awwadi FF, Bardaweel SK. Docking synthesis and anticancer assessment of novel quinoline-amidrazone hybrids. Pharmacia. 2024;71:1-12. doi: 10.3897/pharmacia.71.e117192.
Wang YH, Avonto C, Avula B, Wang M, Rua D, Khan IA. Quantitative determination of α-arbutin β-arbutin kojic acid nicotinamide, hydroquinone resorcinol 4-methoxyphenol 4-ethoxyphenol and ascorbic acid from skin whitening products by HPLC-UV. J AOAC Int. 2015;98(1):5-12. doi: 10.5740/jaoacint.14-123, PMID 25857872.
Neeley E, Fritch G, Fuller A, Wolfe J, Wright J, Flurkey W. Variations in IC(50) values with purity of mushroom tyrosinase. Int J Mol Sci. 2009;10(9):3811-23. doi: 10.3390/ijms10093811, PMID 19865520.
Phasha V, Senabe J, Ndzotoyi P, Okole B, Fouche G, Chuturgoon A. Review on the use of kojic acid a skin-lightening ingredient. Cosmetics. 2022;9(3):64. doi: 10.3390/cosmetics9030064.
Souza PM, Elias ST, Simeoni LA, De Paula JE, Gomes SM, Guerra EN. Plants from Brazilian Cerrado with potent tyrosinase inhibitory activity. PLOS One. 2012;7(11):e48589. doi: 10.1371/journal.pone.0048589, PMID 23173036.
Mukattash HK, Issa R, Abu Hajleh MN, Al Daghistani H. Inhibitory effects of polyphenols from Equisetum ramosissimum and Moringa peregrina extracts on Staphylococcus aureus collagenase and tyrosinase enzymes: in vitro studies. Jordan J Pharm Sci. 2024;17(3):530-48. doi: 10.35516/jjps.v17i3.2164.
Saadh MJ, Ramirez Coronel AA, Saini RS, Arias Gonzales JL, Amin AH, Gavilan JC. Advances in mesenchymal stem/stromal cell-based therapy and their extracellular vesicles for skin wound healing. Hum Cell. 2023;36(4):1253-64. doi: 10.1007/s13577-023-00904-8, PMID 37067766.