
Int J App Pharm, Vol 17, Issue 5, 2025, 534-541Original Article
EXPLORATION OF NICOTINOYL HYDRAZINE DERIVATIVES THROUGH IN SILICO STUDIES FOR NOVEL ANTI-TUBERCULAR DRUG DEVELOPMENT
AKASH HR1, K. ARCHANA DEVI1, S. BELIN1, SENTHIL KUMAR P.1, MOHD ABDUL BAQI2, N. VENKATHESHAN2*, M. SUCHARITHA2, KOPPULA JAYANTHI1*
1Department of Pharmaceutical Chemistry, Arulumigu Kalasalingam College of Pharmacy, Kalasalingam Academy of Research and Education, Krishnan Koil, Tamil Nadu, India. 2Department of Pharmaceutics, Arulumigu Kalasalingam College of Pharmacy, Kalasalingam Academy of Research and Education, Krishnan Koil, Tamil Nadu, India
*Corresponding author: N. Venkatheshan; *Email: jayanthi.k@akcp.ac.in
Received: 22 Apr 2025, Revised and Accepted: 08 Jul 2025
ABSTRACT
Objective: This study explores the interactions of structurally novel nicotinoyl hydrazine derivatives with enoyl-acyl carrier protein reductase (INHA), a key enzyme involved in cellular metabolism regulation. The focus is on the tuberculosis (TB)-associated INHA pathway (PDB ID: 2NSD), aiming to evaluate the anti-tubercular potential of these compounds as inhibitors of the INHA catalytic subunit. Notably, these derivatives, particularly compound 6 (Benzyl nicotinoyl acetamide), feature a unique nicotinoyl hydrazine scaffold with functional groups distinct from known INHA inhibitors such as triclosan or diazaborines.
Methods: Molecular docking of the compounds was performed using the Glide module. An absorption, distribution, metabolism, and excretion (ADME) analysis was conducted using QikProp. The Prime Molecular Mechanics Generalized Born Surface Area (MM-GBSA) method was employed to calculate binding free energy.
Results: Ten compounds exhibited significant binding affinity and interactions, forming hydrogen and pi-cation bonds with key residues such as SER 84, LYS 163, ALA 22, GLY 14, LYS 165, PHE 142, SER 86, TYR 153, LYS 145, TYR 158, MET 167, PHE 168, SER 88, ILE 186, TYR 188, SER 20, PHE 148, and ILE 184 within the INHA catalytic subunit (PDB ID: 2NSD). Among these, Compound 6 demonstrated an exceptional Glide extra precision (XP) docking score of –10.74 kcal/mol and a binding free energy (ΔG_bind) of –109.48 kcal/mol, indicating stronger binding potential than the standard drug isoniazid. Prime MM-GBSA analysis further confirmed its promising binding affinities, with ΔBind (-109.48 kcal/mol), ΔLipo (-45.99 kcal/mol), and ΔVdW (-73.62 kcal/mol). The ligand consistently interacted with residues SER 88, SER 86, ILE 186, and TYR 188.
Conclusion: Benzyl nicotinoyl acetamide (Compound 6), with its structurally novel scaffold and potent binding metrics, exhibits significant potential as an INHA inhibitor. Characterized by functional groups such as a secondary amine (NH), ether (C-O-C), hydroxyl (OH), pyridine, carbonyl (C=O), and phenyl (C6H5), it shows potential application as an anti-tubercular agent, particularly for tuberculosis treatment.
Keywords: Enoyl-Acyl carrier protein reductase (INHA), Molecular docking, MM-GBSA, ADME, Tuberculosis, Isoniazid, Anti-tubercular agents
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i5.54697 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Tuberculosis (TB), caused by Mycobacterium tuberculosis (MTB), continues to be one of the most devastating infectious diseases globally. It is estimated that approximately one-third of the world’s population is infected with latent TB, as indicated by a positive tuberculin skin test. Although not all infected individuals develop active disease, TB remains a significant public health challenge, particularly in 22 high-burden countries that collectively account for over 80% of global TB cases. TB primarily affects the lungs (pulmonary TB), but it can also involve other organs (extrapulmonary TB). Transmission occurs through airborne particles released during coughing, sneezing, or talking by infected individuals [1]. MTB is an aerobic, non-motile, non–non-spore-forming bacillus characterized by its acid-fast staining, due to a high lipid content in its cell wall, which also contributes to its virulence and immune evasion capabilities. One of the major challenges in controlling TB is the bacterium's slow growth rate, with a generation time of about 20 h. Cultures often require 3 to 8 w to show visible growth, delaying diagnosis and treatment. MTB is an intracellular pathogen that can persist in a dormant state within host cells, reactivating later under favorable conditions. However, the molecular mechanisms governing dormancy and reactivation remain poorly understood, representing a critical area for ongoing research [2]. TB predominantly affects adults and shows a higher prevalence among men compared to women. In 2019, nearly 10 million people were diagnosed with TB, resulting in approximately 1.4 million deaths globally. The burden is particularly high in South-East Asia, Africa, and the Western Pacific regions. Moreover, the COVID-19 pandemic has disrupted TB services, causing a rise in TB-related deaths between 2019 and 2021 [3]. Enoyl-acyl carrier protein reductase (INHA) plays a pivotal role in the fatty acid elongation cycle involved in mycolic acid biosynthesis, a critical component of the Mycobacterium tuberculosis cell wall that confers structural integrity and resistance to host defences. Disruption of INHA impairs cell wall synthesis, leading to bacterial death, which has established it as a well-validated target, particularly in drug-resistant strains. In contrast to catalase-peroxidase (KatG), which indirectly affects INHA by activating pro-drugs like isoniazid, direct inhibition of INHA bypasses the dependency on enzymatic activation-an important consideration in KatG mutation–mediated resistance. Similarly, RNA polymerase beta subunit gene the target of rifampicin, is often mutated in multidrug-resistant tuberculosis (MDR-TB) cases. Thus, INHA offers a more direct and potentially resistance-resilient target, making it highly suitable for novel inhibitor development. A notable decline in TB incidence has occurred in some countries, but others continue to experience rising or stagnant rates. Risk factors such as hsuman immunodeficiency viruses (HIV) infection, diabetes, undernutrition, smoking, alcohol use, and exposure to environmental pollutants significantly increase susceptibility to TB. Social determinants like poverty, overcrowding, and limited literacy also play a crucial role in disease transmission and outcomes. The emergence of multidrug-resistant (MDR-TB) and extensively drug-resistant TB (XDR-TB) has further complicated treatment efforts. Several new and repurposed drugs are under investigation or have been introduced to combat resistant TB strains. Bedaquiline, a diarylquinoline, is now recommended for rifampicin-resistant TB. Other agents, such as delamanid, pretomanid, and PA-824, are being evaluated in combination regimens for enhanced efficacy against resistant TB [4]. Isoniazid (INHA), a cornerstone of TB treatment, is known to cause toxicity, especially in individuals with pyridoxine deficiency. INH interferes with pyridoxine metabolism in two ways: by inhibiting pyridoxine phosphokinase and by forming an inactive complex with pyridoxal phosphate, which can result in neurological side effects like seizures or neuropathy due to decreased gamma-aminobutyric acid (GABA) synthesis. Additionally, INH-induced hepatotoxicity is associated with toxic metabolites such as hydrazine and acetyl hydrazine. Patients with a slow acetylator phenotype, or those taking hepatotoxic drugs like rifampin or pyrazinamide, are at heightened risk [5]. In summary, TB remains a pressing global health concern due to its widespread prevalence, slow diagnosis, and treatment challenges, including drug resistance and toxicity. Enhanced research into host-pathogen interactions, novel therapeutics, and public health interventions is essential to control and eventually eliminate the tuberculosis disease.
MATERIALS AND METHODS
Materials
The Schrödinger Suite was employed for computational studies, including molecular docking, MM-GBSA calculations, and ADME property predictions. The INHA enzyme complexed with its ligand (PDB ID: 2NSD) was retrieved from the Protein Data Bank for docking simulations. Ligand structures, derived from novel 2-(4-(2-nicotinoylhydrazineyl) phenoxy)-N-phenylacetamide analogs, were either obtained from existing sources or generated for the study.
Molecular docking
Molecular docking was employed to predict the binding affinities and interaction mechanisms between ligands and INHA. The objective was to identify the best docked conformations based on e-model, energy, and score values. Schrödinger Suite 2021-4 was utilized to prepare the X-ray crystal structure of INHA (PDB ID: 2NSD, Resolution 1.90 Å resolution) [6]. This preparation involved adding hydrogen atoms, optimizing protonation states, and ensuring structural readiness for docking (Schrödinger, 2021-4). Crystallographic water molecules were removed to prevent interfering interactions, and missing side chains were reconstructed using the Prime module [7]. Ligand structures were processed using LigPrep, generating multiple conformations and tautomers for ten compounds [8]. Docking simulations were conducted with the optimized potentials for liquid simulations (OPLS)3-2005 force field, recognized for its accuracy in modeling non-covalent interactions while maintaining computational efficiency [9]. To validate the docking protocol, the co-crystallized ligand from PDB ID: 2NSD was redocked into the binding site using the same Glide XP parameters. The root mean square deviation (RMSD) between the experimental and redocked poses was 1.8 Å, indicating high reliability of the docking method in reproducing known binding orientations. The active site was defined using a 20 Å grid box centered at the grid center defined at the coordinates X = 35.21, Y = 18.43, Z = 44.87. on the co-crystallized ligand in fig. 1, which facilitated precise docking calculations [10, 11]. Glide XP was employed for docking, providing an in-depth evaluation of ligand binding conformations. The most favorable docked conformations were selected based on Glide energy, score, and e-model values (Schrödinger, 2023-4) [12]. The resulting protein-ligand complexes were analyzed and visualized to examine their interactions and conformations, as depicted in fig. 2.
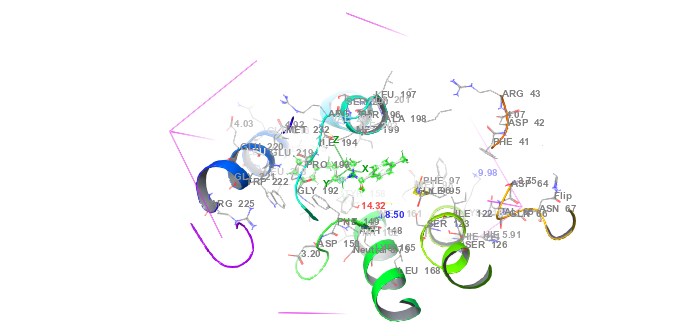
Fig. 1: Protein-ligand interaction grid complex (PDB id: 2NSD) in molecular docking
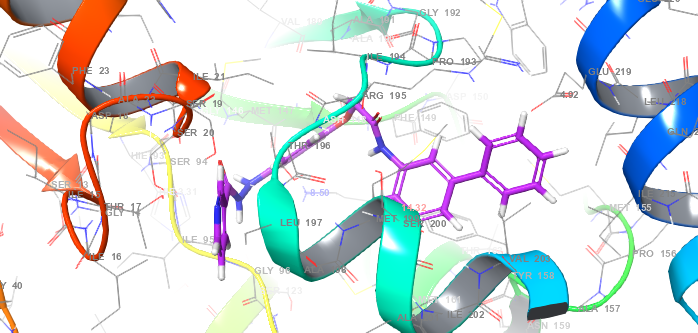
Fig. 2: Protein-ligand interaction complex (PDB id: 2NSD) in molecular docking
Binding free energy calculations using prime MM-GBSA
The binding free energy of each protein-ligand complex was determined using the Prime MM-GBSA technique in Schrödinger Suite 2021-4. This method provides a comprehensive assessment of binding affinity by integrating various energy contributions. Prime MM-GBSA calculates binding free energy by combining implicit solvation models with molecular mechanics energies. The process involves several key steps, including energy minimization of each protein-ligand complex using the OPLS3e force field, which is specifically designed for high-accuracy modeling of biomolecular interactions. To account for solvation effects, the variable dielectric generalized born (VSGB) 2.0 implicit solvation model was applied, offering a detailed representation of hydrogen bonding, self-contact interactions, and hydrophobic effects. The MM-GBSA method determines binding free energy by summing three major components: Surface Area Term (representing hydrophobic effects), Generalized Born Solvation Energy (accounting for implicit solvation), and Molecular Mechanics Energy (capturing van der Waals and electrostatic interactions). The binding energy is estimated by subtracting the total free energies of the individual protein and ligand from the free energy of the protein-ligand complex [13]. This calculation provides insights into the stability and strength of ligand-target interactions. Additionally, the MM-GBSA approach incorporates physics-based corrections to improve accuracy, addressing interaction effects that may not be fully captured by standard energy terms. These refinements enhance the reliability of binding affinity predictions, offering a more precise evaluation of ligand-protein interactions.
ADME calculations
The pharmacokinetic parameters-Absorption, Distribution, Metabolism, and Excretion (ADME)-for the protein-ligand complexes were predicted using Schrödinger Suite 2021-4. Protein structures were either obtained from experimentally resolved data or generated through homology modeling, while ligand molecules were prepared using standard molecular modeling protocols. The QikProp module of the Prime tool in Schrödinger was employed for ADME prediction [14]. To ensure accurate modeling of molecular interactions, the OPLS3-2005 force field was used, which is a refined version of the Optimized Potentials for Liquid Simulations (OPLS3e) force field. It is well-recognized for its enhanced performance in predicting protein-ligand behavior and molecular properties. The solvation environment was modelled using the VSGB 2.0 solvation model, which considers the dynamic nature of solvents in biological systems [15]. Both proteins and ligands were pre-processed using Schrödinger’s preparation tools, which included assigning appropriate protonation states at physiological pH and performing energy minimization. The complexes were further minimized using the OPLS3-2005 force field to obtain low-energy conformations. ADME parameters such as permeability, oral absorption, metabolism, and excretion were then predicted using QikProp, providing reliable estimates based on validated empirical models.
Compounds used
The study focused on a series of chemical structures derived from the nicotinoyl hydrazine scaffold, known for its anti-tuberculosis activity. These compounds are structurally related to the well-established anti-tubercular drug Isoniazid and are designed to selectively activate the INHA enzyme, a key target in Mycobacterium tuberculosis. The synthesized derivatives incorporated both polar substituents—such as Cl, NO₂, NH₂, OH, CN, and F—and non-polar groups—like CH₃, C₆H₅, and Br. These functional groups were selected based on their potential to enhance interaction with the INHA enzyme, thereby improving the inhibitory activity. The presence of these moieties is believed to increase the likelihood of enzyme activation or binding, offering promising therapeutic potential for the treatment of tuberculosis.
RESULTS
Docking results and analysis
Molecular docking studies were performed using the crystal structure of INHA from Mycobacterium tuberculosis (PDB ID: 2NSD) within the Schrödinger Suite 2021-4 platform. Ligand conformations were validated through virtual screening, ensuring a root mean square deviation (RMSD) of less than 1.8 Å from the native co-crystallized ligand, confirming accurate pose prediction. To ensure drug-likeness and eliminate compounds with unfavorable pharmacokinetic profiles, Lipinski’s Rule of Five was applied during the selection process. The docking performance of ligands was evaluated using multiple Glide XP metrics, including the Glide score, E-model, van der Waals energy (E_vdw), Coulombic energy (E_coul), and the overall binding energy (Energy). These parameters collectively provided insights into binding affinity, molecular interactions, and the stability of the protein-ligand complexes.
Table 1: The XP-docking scores for compounds 1–10 in INHA catalytic pocket (PDB ID: 2NSD)
| Compound | aGscore | bGvedw | cGecou | eGenergy | fGemodel |
| 1 | -7.83 | -45.53 | -3.82 | -49.35 | -79.48 |
| 2 | -5.87 | -52.77 | -5.76 | -58.54 | -90.82 |
| 3 | -7.22 | -47.28 | -3.36 | -50.65 | -82.98 |
| 4 | -7.68 | -50.35 | -8.63 | -58.98 | -91.91 |
| 5 | -7.35 | -48.40 | -7.80 | -56.20 | -98.08 |
| 6 | -10.74 | -56.76 | -6.32 | -63.08 | -109.52 |
| 7 | -7.20 | -49.31 | -7.36 | -56.67 | -84.39 |
| 8 | -7.87 | -50.5 | -8.09 | -58.59 | -85.16 |
| 9 | -8.52 | -39.39 | -4.66 | -44.05 | -77.32 |
| 10 | -6.68 | -52.20 | -5.85 | -58.06 | -92.78 |
| Co-crystal | -10.17 | -38.53 | -3.88 | -42.41 | -63.15 |
| Standard | -6.04 | -18.59 | -4.65 | -23.24 | -29.48 |
aGlide Score, bGlide Van der Waals Energy, cGlide Coulomb Energy, dGlide Energy. fGlide E-model
The docking analysis revealed that all synthesized compounds exhibited favorable binding affinities toward the INHA enzyme when compared to the standard anti-tubercular drug Isoniazid. Among the tested molecules, compounds 6, 9, and 8 demonstrated the highest Glide scores of –10.74 kcal/mol, –8.52 kcal/mol, and –7.87 kcal/mol, respectively, indicating strong and stable binding interactions. Compounds 1, 4, and 5 followed closely with Glide scores of –7.83 kcal/mol, –7.68 kcal/mol, and –7.35 kcal/mol, respectively, suggesting good binding affinity. In comparison, compounds 3, 7, and 10 showed slightly lower Glide scores of –7.22 kcal/mol, –7.20 kcal/mol, and –6.68 kcal/mol, but still maintained notable interaction potential. Conversely, compound 2 displayed the lowest binding affinity with a Glide score of –5.87 kcal/mol. The standard drug Isoniazid showed a Glide score of –6.04 kcal/mol, serving as a benchmark for comparison. Notably, compounds 6, 9, and 8 also exhibited favorable interaction energies, including van der Waals energy (E_vdw) of –56.76, –39.39, and –50.50 kcal/mol, Coulombic energy (E_coul) of –6.32, –4.66, and –8.09 kcal/mol, and total docking energies (E_energy) of –63.08, –44.05, and –58.59 kcal/mol, respectively. The E-model scores for these compounds were –109.52, –77.32, and –85.16 kcal/mol, further supporting their strong binding efficiency.
These findings suggest that compounds 6, 9, and 8 are the most promising candidates for INHA enzyme inhibition, with binding affinities comparable to or even exceeding that of Isoniazid, thus warranting further investigation.
Binding free energy contributions using MM-GBSA
The binding free energy (ΔG_bind) for each compound complexed with INHA (PDB ID: 2NSD) was calculated using the MM-GBSA method. The results are summarized in table 2. The total ΔG_bind values were derived from individual energy components, including Coulombic interactions (ΔG_Coul), hydrophobic (lipophilic) contributions (ΔG_Lip), hydrogen bonding energy (ΔG_HB), and van der Waals interactions (ΔG_VdW). These components together reflect the overall thermodynamic stability and binding strength of each ligand within the active site of the INHA enzyme.
Table 2: Binding free energy (MM-GBSA) contribution (kcal/mol) for compounds 1–10 in INHA complexes
| Compound | GBind | GCoul | GCov | GHB | GLipo | GSolv | Gvdw | Genergy |
| 1 | -91.21 | -21.86 | 15.01 | -2.96 | -40.69 | 44.79 | -84.38 | 6.39 |
| 2 | -62.71 | -31.11 | 7.77 | -3.44 | -28.40 | 49.39 | -56.00 | 4.69 |
| 3 | -95.11 | 31.51 | 10.77 | -2.70 | -41.05 | 59.85 | -87.69 | 7.38 |
| 4 | -113.71 | 19.86 | 27.63 | -3.27 | -47.26 | 31.70 | -100.6 | 3.91 |
| 5 | -112.02 | -18.73 | 14.21 | -2.26 | -48.47 | 28.15 | -83.79 | 8.60 |
| 6 | -109.48 | -34.41 | 3.17 | -4.08 | -45.99 | 48.46 | -73.62 | 5.87 |
| 7 | -82.76 | -59.57 | 7.43 | -4.10 | -34.48 | 69.30 | -61.15 | 4.79 |
| 8 | -67.01 | 43.30 | 7.82 | -3.39 | -26.51 | 48.75 | -49.16 | 4.87 |
| 9 | -59.53 | -9.80 | 6.52 | -0.58 | -31.89 | 21.11 | -45.69 | 6.38 |
| 10 | -99.41 | -46.79 | 16.29 | -5.91 | -30.06 | 31.24 | -62.64 | 4.76 |
| Co-crystal | -63.25 | -26.69 | 3.54 | -1.48 | -33.41 | 32.41 | -37.64 | 1.38 |
| Standard | -16.25 | -27.51 | 4.48 | -3.18 | -8.48 | 30.98 | -13.96 | 0.58 |
aFree Energy of Binding, bCoulomb Energy, ccovalentd Hydrogen Bonding Energy, eHydrophobic Energy (non-polar contribution estimated by solvent accessible surface area),f solvent gVan der Waals Energy.
The MM-GBSA analysis revealed that compound 4 exhibited the most favorable binding free energy (ΔG_bind) of –113.71 kcal/mol, with significant contributions from Coulombic energy (–19.86 kcal/mol), hydrophobic energy (ΔG_Lipo: –47.26 kcal/mol), and van der Waals interactions (ΔG_VdW: –100.63 kcal/mol). Similarly, compound 5 showed a strong binding free energy of –112.02 kcal/mol, driven by Coulombic (–18.73 kcal/mol) and hydrophobic (–48.47 kcal/mol) interactions. Compound 6 also demonstrated notable binding characteristics, with a ΔG_bind of –109.48 kcal/mol, supported by Coulombic energy (–34.41 kcal/mol), hydrophobic energy (–45.99 kcal/mol), and van der Waals energy (–73.62 kcal/mol). Compound 10 followed with a binding free energy of –99.41 kcal/mol, primarily due to Coulombic (–46.79 kcal/mol), hydrophobic (–30.06 kcal/mol), and van der Waals (–62.64 kcal/mol) contributions. Compound 3 recorded a ΔG_bind of –95.11 kcal/mol, with major energy contributions from Coulombic (–31.51 kcal/mol) and hydrophobic (–41.05 kcal/mol) interactions. Compound 1 had a binding energy of –91.21 kcal/mol, driven by Coulombic (–21.86 kcal/mol), hydrophobic (–40.69 kcal/mol), and van der Waals (–84.38 kcal/mol) terms. Lower binding affinities were observed for compounds 7, 8, and 2, with ΔG_bind values of –82.76 kcal/mol, –67.01 kcal/mol, and –62.71 kcal/mol, respectively. These compounds showed relatively lower Coulombic (–59.57, –43.30, –31.11 kcal/mol) and hydrophobic energy (–34.48, –26.51, –28.04 kcal/mol) contributions. In contrast, the standard drug Isoniazid exhibited a binding free energy of only –16.23 kcal/mol, indicating a significantly lower binding potential compared to the synthesized compounds. This highlights the enhanced interaction capabilities of the designed molecules with the INHA target.
Hydrogen bonding and amino acid interactions
Table 3 provides a detailed summary of the hydrogen bonding interactions formed between each compound and the amino acid residues within the catalytic pocket of INHA, PDB ID: 2NSD. These interactions play a critical role in determining the binding affinity, stability, and inhibitory potential of the protein-ligand complexes. The presence of strong and specific hydrogen bonds significantly enhances the overall molecular interaction profile, supporting the compounds’ potential as effective INHA inhibitors.
Table 3: Number of hydrogen bonds and specific amino acid residues involved in compound interactions within the INHA catalytic pocket (PDB ID: 2NSD)
| Compound | H-bond | Pi-Pi stacking | Amino acids |
| 1 | 1 | 0 | SER 84 |
| 2 | 3 | 0 | LYS 163, ALA 22, GLY 14 |
| 3 | 2 | 1 | GLY 165, PHE 142 |
| 4 | 4 | 1 | SER 86, TYR 153, GLY 145, PHE 142 |
| 5 | 5 | 2 | SER 86, PHE 168, TYR 158, MET 167, |
| 6 | 4 | 1 | SER 88, SER 86, ILE 186, TYR 188 |
| 7 | 2 | 0 | TYR 188, SER 20 |
| 8 | 2 | 0 | TYR 158, SER 20 |
| 9 | 2 | 1 | PHE 148, TYR 153 |
| 10 | 4 | 1 | ILE 184, TYR 153, SER 86, SER 20 |
| 11 | 1 | 0 | TYR 158 |
| 12 | 2 | 0 | ILE 194, TYR 158 |
Each compound's hydrogen bond count and interacting amino acid residues are included in the table.
Table 3 summarizes the hydrogen bond interactions between each compound and the catalytic pocket of INHA (PDB ID: 2NSD). Most compounds demonstrated strong hydrogen bonding with key amino acid residues, reinforcing their potential inhibitory activity. Notably, compound 5 exhibited the highest number of hydrogen bonds (five), interacting with critical residues such as SER86, TYR158, PHE168, SER86, and MET167, highlighting its strong binding affinity and potential as a potent INHA inhibitor. Compounds 6, 4, and 10 each formed four hydrogen bonds with key residues like SER88, ILE186, TYR182, PHE142, LYS145, and ILE184, reflecting considerable interaction strength. Compound 2 established three hydrogen bonds, engaging residues such as LYS163, ALA22, and GLY14, while compounds 3, 7, 8, and 9 formed two hydrogen bonds each with residues including LYS165, PHE142, TYR158, SER20, PHE148, and TYR158. Although these compounds formed fewer hydrogen bonds, they still maintained meaningful interactions within the catalytic pocket. For comparison, the standard drug Isoniazid formed two hydrogen bonds with ILE194 and TYR158, indicating a moderate level of interaction. Several synthesized compounds showed stronger or comparable binding interactions, suggesting improved inhibitory potential over the standard treatment.
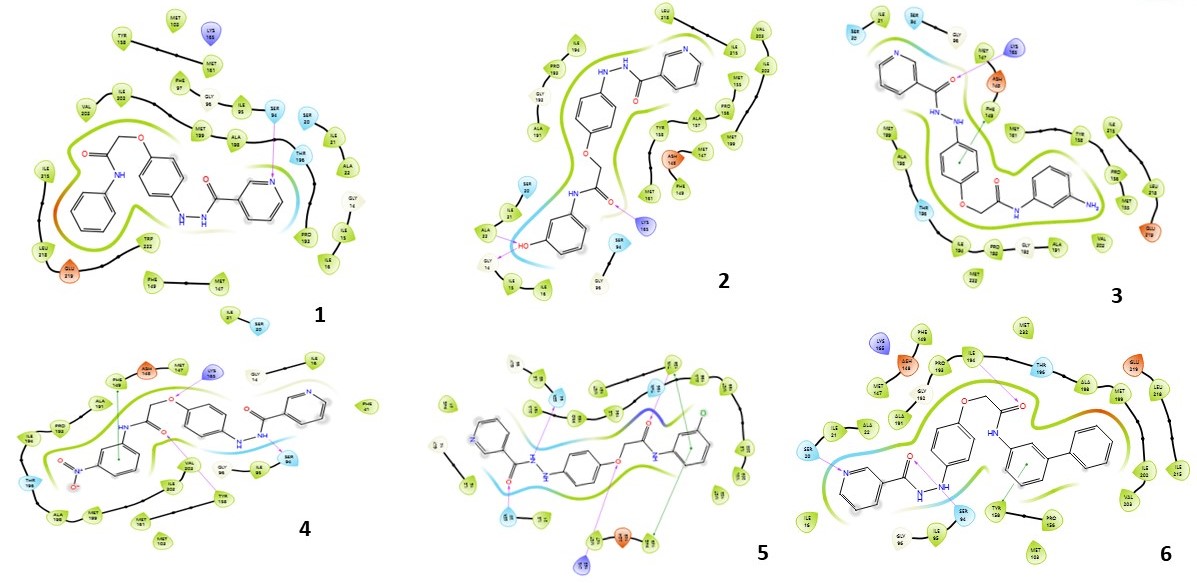
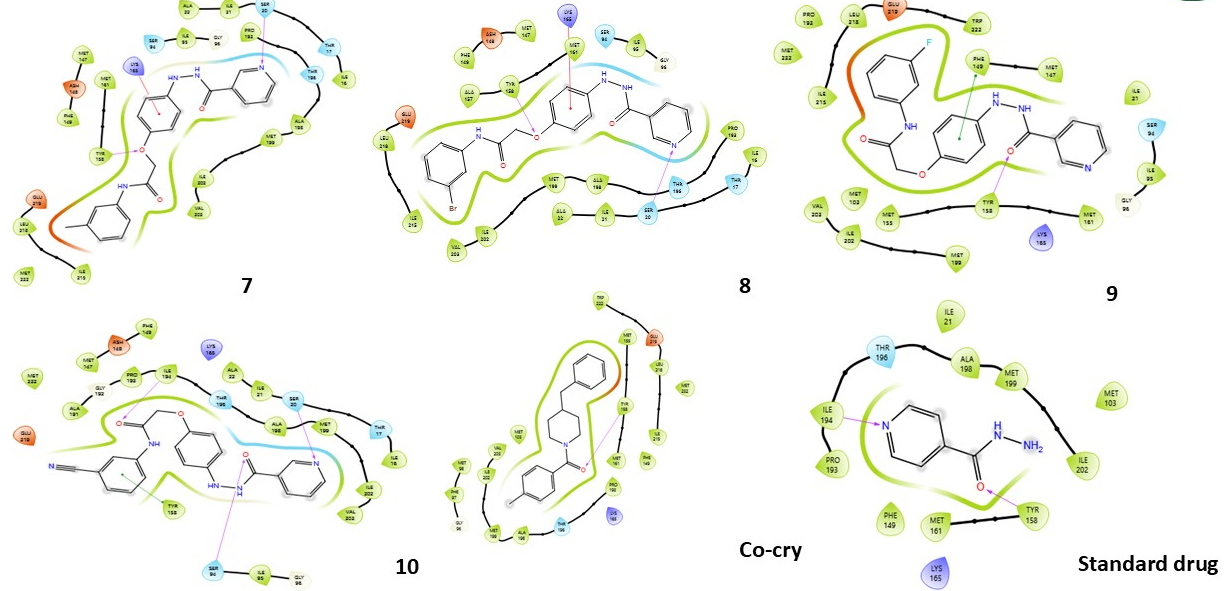
Fig. 3: Compound 1-10 with standard drug 2D interaction diagrams in the INHA catalytic pocket PDB ID: (2NSD)
Fig. 3 presents the 2D interaction diagrams of the ten studied compounds, highlighting their binding interactions within the catalytic pocket of INHA (PDB ID: 2NSD). The diagrams depict key interactions, including hydrogen bonds formed through functional groups such as carbonyl (C=O), amine (NH), hydroxyl (OH), ether linkages (C–O–C), oxygen atoms (O), and nitrogen in pyridine rings (N). These groups establish crucial hydrogen bonds with specific amino acid residues, contributing to the stabilization of the ligand–receptor complex. Additionally, hydrophobic interactions, particularly involving aromatic rings, are evident. Notably, π–cation interactions are observed between aromatic residues in the receptor and the aromatic moieties of the ligands. Together, these visual representations in fig. 3 offer a clear and insightful understanding of how distinct functional groups contribute to effective INHA binding while also identifying the key interacting residues.
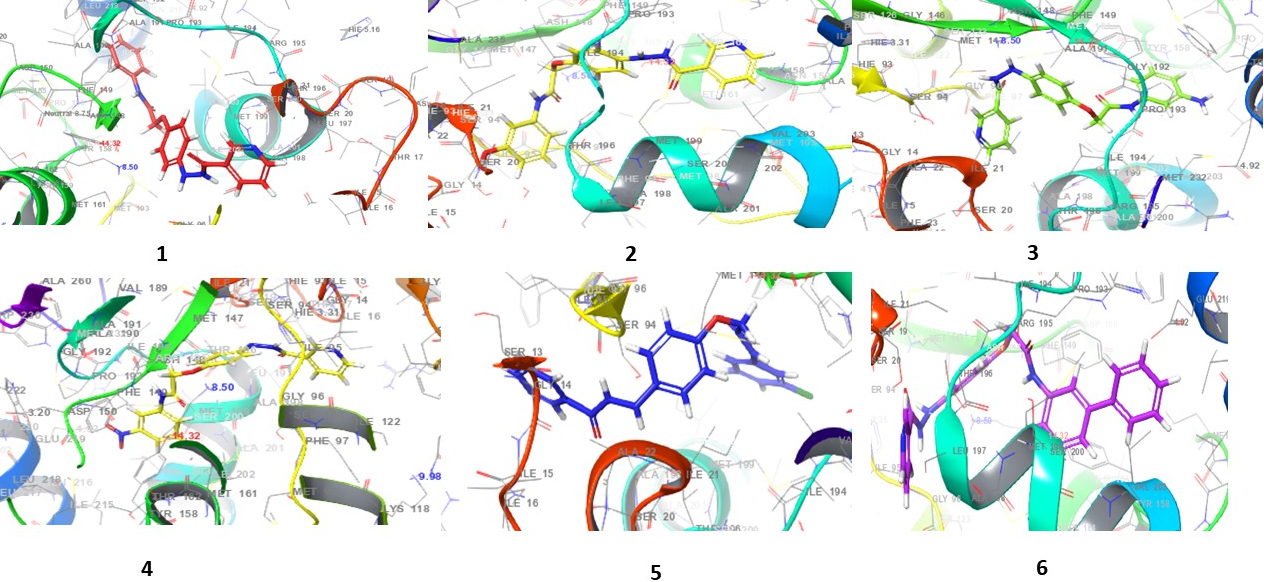
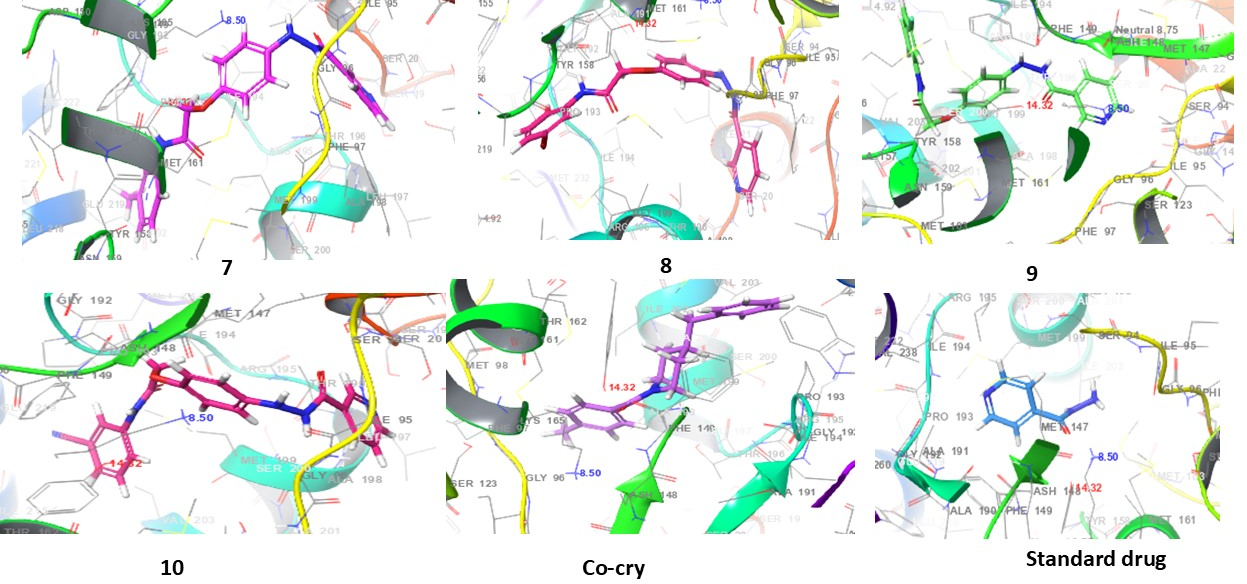
Fig. 4: 3D interaction diagrams of compounds (1-10) and standard drug in the INHA catalytic pocket PDB ID: (2NSD)
Fig. 4 presents the 3D interaction diagrams of the ten compounds within the INHA catalytic pocket (PDB ID: 2NSD). These diagrams depict the spatial orientation and binding conformation of each compound within the active site of the receptor. The visualizations highlight key interactions between the compounds' functional groups-such as carbonyls (C=O), amines (NH), oxygen-containing groups (O), pyridine rings (N), ether linkages (C–O–C), and hydroxyls (OH)-and the amino acid residues of the receptor. These interactions include hydrogen bonding and hydrophobic contacts, particularly involving aromatic rings. Additionally, π-cation interactions are observed between aromatic residues in the receptor and the aromatic moieties of the ligands. This 3D representation offers valuable insights into the nature of molecular contacts and the stability of the ligand–receptor complexes, shedding light on how these interactions contribute to binding affinity and receptor function.
ADME study results
The ADME properties of the ten compounds were analyzed to evaluate their pharmacokinetic behavior and safety profiles. A summary of the results is provided in table 4, followed by a detailed interpretation of the findings.
The ADME analysis indicates that all ten compounds exhibit minimal central nervous system (CNS) penetration, with values ≤-2, suggesting a low risk of CNS-related side effects. The Solvent Accessible Surface Area (SASA) values range from 789.74 to 626.66 Ų, reflecting favorable surface interactions and potential for effective absorption. The compounds possess 3to 4.5 hydrogen bond donors and 5.25 to 6.75 hydrogen bond acceptors, indicating optimal hydrogen bonding capability for target interaction. Lipophilicity (QPlog P) values range between 2.75 and 5.29, suggesting a balanced profile for membrane permeability and solubility. Caco-2 permeability values span from 158.38 to 1408.78 nm/s, where higher values imply enhanced intestinal absorption. The cardiotoxicity risk, measured by QPlog HERG, remains low across all compounds, with values ranging from -7.58 to -5.41. Additionally, negative QPlog BB values (ranging from -1.95 to 0.73) further support a low potential for blood-brain barrier penetration. Most compounds meet Lipinski’s Rule of Five criteria, indicating good oral bioavailability. Collectively, these ADME properties suggest that the compounds are promising candidates for further development as safe and effective therapeutic agents.
Comparative analysis of compounds with standard drug
A comparative analysis of ten compounds targeting INHA, benchmarked against the standard drug isoniazid, highlights their promising therapeutic potential. Molecular docking studies reveal that compounds 1, 3, 4, 5, 6, 7, 8, 9, and 10 exhibit strong binding affinities ranging from-6.68 to-10.74 kcal/mol, all outperforming isoniazid, which shows a binding affinity of-6.04 kcal/mol. Notably, compound 6 demonstrates the highest affinity at-10.74 kcal/mol. Further validation using MMGBSA calculations confirms robust binding free energies for all ten compounds, with values ranging from-59.53 to-113.71 kcal/mol, significantly surpassing the standard drug’s energy of-16.23 kcal/mol. Compound 4 exhibits the strongest binding energy at-113.71 kcal/mol. Interaction analysis reveals that these compounds form up to five hydrogen bonds with key INHA residues, compared to only two formed by isoniazid, indicating stronger and more stable binding. Additionally, ADME profiling shows minimal CNS penetration, low cardiotoxicity risk, and favorable oral absorption and lipophilicity across all compounds. Despite minor differences in hydrogen bonding and energy values, each compound demonstrates superior activity compared to isoniazid, with compound 6 emerging as a particularly promising INHA inhibitor for further development.
Table 4: ADME properties of compounds 1-10 and standard drug
| Comp | CNS | SASA | Donar HB | Accept HB | Qplog Po/w | Qplog HERG | QPP Caco |
QPlogBB | Human oral absorption | PSA | Rule of five |
| 1 | -2 | 690.72 | 3 | 8.25 | 2.49 | -7.51 | 364.49 | -1.52 | 3 | 107.19 | 0 |
| 2 | -2 | 701.11 | 4 | 9 | 1.86 | -7.32 | 147.72 | -2.04 | 3 | 128.24 | 0 |
| 3 | -2 | 663.33 | 4.5 | 9.25 | 1.34 | -6.62 | 98.26 | -2.08 | 3 | 131.27 | 0 |
| 4 | -2 | 721.58 | 3 | 9.25 | 1.85 | -7.25 | 54.22 | -2.56 | 3 | 151.42 | 0 |
| 5 | -2 | 710.28 | 3 | 8.25 | 3.07 | -7.29 | 488.21 | -1.20 | 3 | 106.79 | 0 |
| 6 | -2 | 783.56 | 3 | 8.25 | 3.91 | -8.12 | 431.53 | -1.54 | 3 | 103.6 | 0 |
| 7 | -2 | 723.55 | 3 | 8.25 | 2.83 | -7.33 | 357.10 | -1.55 | 3 | 109.37 | 0 |
| 8 | -2 | 719.55 | 3 | 8.25 | 3.11 | -7.34 | 369.67 | -1.34 | 3 | 109.47 | 0 |
| 9 | -2 | 666.16 | 3 | 8.25 | 2.59 | -6.77 | 468.87 | -1.19 | 3 | 101.02 | 0 |
| 10 | -2 | 698.68 | 3 | 9.75 | 1.71 | -6.98 | 90.73 | -2.20 | 3 | 131.97 | 0 |
| Co-crystal | 1 | 613.31 | 0 | 3 | 4.90 | -5.92 | 4308.68 | 0.008 | 3 | 28.69 | |
| Standard | -1 | 333.26 | 3 | 4.5 | 0.64 | -3.64 | 264.13 | 0.86 | 2 | 82.09 | 0 |
CNS: Central Nervous System Penetration (values ≤-2 indicate low CNS penetration). SASA: Solvent Accessible Surface Area (in Ų), indicative of molecular surface interaction. Donor HB/Acceptor HB: Number of hydrogen bond donors and acceptors. QPlog P o/w: Octanol-water partition coefficient, indicating lipophilicity. QP Caco: Permeability across Caco-2 cell monolayers (nm/s), reflecting intestinal absorption. QPlog HERG: Potential for interaction with the HERG channel (negative values indicate a lower risk of cardiotoxicity). PSA: Polar Surface Area (in Ų), affecting drug permeability. QPlog BB: Blood-brain barrier permeability (negative values indicate low permeability). Human Oral Absorption: Predicted oral absorption potential. Rule of Five: Compliance with Lipinski's Rule of Five.
DISCUSSION
The molecular docking and ADME analyses highlight the therapeutic promise of benzyl nicotinoyl acetamide derivatives, particularly compound-6 (benzyl nicotinoyl acetamide), as potent inhibitors of INHA—a crucial enzyme involved in mycolic acid biosynthesis and cell wall formation in Mycobacterium tuberculosis [16]. Compound-6exhibited a superior docking score of-10.74 kcal/mol, outperforming the standard anti-tubercular drug isoniazid (-6.04 kcal/mol) [17]. Moreover, the binding free energy of-109.48 kcal/mol [18], further supports the stability and strength of the ligand–target interaction. This compound forms multiple hydrogen bonds with key INHA active site residues such as SER86, SER88, ILE186, and TYR188, indicating strong and specific inhibitory potential. ADME profiling of the compound suggests good oral bioavailability, low CNS penetration, and minimal cardiotoxicity, reinforcing its systemic safety profile. These findings suggest that compound-6 may offer a promising alternative or adjunct to current anti-TB therapies, with selective INHA inhibition and limited off-target effects. Comprehensive toxicity studies and extended ADME evaluations will also be necessary to confirm the safety and drug-like nature of these derivatives. Overall, the development of benzyl nicotinoyl acetamide derivatives—especially compound-6—offers a novel and targeted strategy for tuberculosis treatment [19]. With continued research and validation, these compounds may contribute significantly to the pipeline of next-generation anti-TB agents, ultimately benefiting global public health. Further in vitro and in vivo studies in tuberculosis models are essential to establish the anti-mycobacterial efficacy, optimal dosage, and pharmacological effects.
To the best of our knowledge, nicotinoyl acetamide derivatives-particularly compound-6 (benzyl nicotinoyl acetamide)-have not been previously investigated as potential inhibitors of INHA. Comprehensive literature reviews and database searches using platforms such as PubChem, ChEMBL, and DrugBank confirm the novelty of these compounds in both structure and biological application. No prior studies have reported their association with INHA inhibition, making this investigation the first to highlight their potential in this context. In this study, compound-6 exhibited strong binding affinity and favorable pharmacokinetic properties, underscoring its promise as a lead molecule against INHA, a well-validated target in the treatment of tuberculosis. The observed molecular interactions, along with predicted low toxicity and good oral bioavailability, suggest that these derivatives could serve as a new class of anti-tubercular agents. If further validated through experimental methods and clinical research, these novel molecules may offer significant therapeutic advantages, especially for addressing drug-resistant strains of Mycobacterium tuberculosis. Resistance to isoniazid, one of the frontline anti-TB agents, frequently arises due to mutations in the INHA promoter region, resulting in increased expression of the INHA enzyme and reduced susceptibility to inhibition. Other common resistance mechanisms include mutations in the KatG gene, which encodes a catalase-peroxidase required for isoniazid activation. These genetic alterations render isoniazid less effective, highlighting the critical need for direct INHA inhibitors that can maintain efficacy even in the presence of such mutations. Novel compounds that do not rely on enzymatic activation and that bind strongly to INHA—such as the nicotinoyl hydrazine derivatives explored in this study—represent a promising strategy to overcome existing resistance pathways and expand treatment options for MDR and XDR-TB. Additionally, their involvement in fatty acid biosynthesis suggests potential relevance to broader metabolic pathways, presenting opportunities for multifunctional drug development in the future. Our findings show that compound 6 exhibits stronger binding affinity (ΔG_bind = –109.48 kcal/mol) than previously reported INHA inhibitors such as isoniazid and diazaborine analogs, which generally exhibit lower binding energies (~ –70 to –90 kcal/mol) [20]. These results emphasize the enhanced potential of our nicotinoyl hydrazine derivatives as promising anti-tubercular agents.
CONCLUSION
The molecular docking and ADME analyses highlight the therapeutic potential of benzyl nicotinoyl acetamide derivatives, particularly compound-6 (benzyl nicotinoyl acetamide), as promising inhibitors of INHA—a validated target in antitubercular therapy. Compound-6 demonstrated strong binding affinity and stable interactions through multiple hydrogen bonds with critical INHA residues, including SER88, SER86, ILE186, and TYR188, indicating its potential as a robust INHA inhibitor. ADME profiling further supports its drug-likeness, with favorable properties such as good oral bioavailability, minimal CNS penetration, and low cardiotoxicity, suggesting its suitability for systemic use with a safer profile than conventional anti-tubercularagents.
To confirm its therapeutic efficacy, future research should prioritize experimental validation using techniques like Surface Plasmon Resonance (SPR), Isothermal Titration Calorimetry (ITC), and molecular dynamics simulations to assess the interaction stability over time. Additionally, comprehensive in vitro and in vivo evaluations in tuberculosis models, along with detailed toxicity and extended ADME studies, will be crucial for ensuring safety and clinical feasibility. Overall, the integration of benzyl nicotinoyl acetamide derivatives into tuberculosis treatment regimens represents a novel and targeted approach, potentially offering safer and more effective alternatives, especially in addressing drug-resistant Mycobacterium tuberculosis infections.
ACKNOWLEDGMENT
The authors would like to thank the Department of Pharmaceutical Chemistry, Arulumigu Kalasalingam College of Pharmacy, Virudhunagar, Tamil Nadu, for providing facilities for conducting Research.
FUNDING
No funding was received for this work.
AUTHORS CONTRIBUTIONS
Dr. Koppula Jayanthi-Conceptualization, validation, original draft preparation, and data curation, Dr. Mohd Abdul Baqi-Data curation and Dr. Narayana Venkateswaran-Review and editing, Akash HR, Archana Devi, S. Belin, Senthil Kumar. P and M. Sucharitha-Data curation, methodology, and writing review.
CONFLICT OF INTERESTS
Declared none
REFERENCES
Zhang X, Zhao R, Qi Y, Yan X, Qi G, Peng Q. The progress of mycobacterium tuberculosis drug targets. Front Med (Lausanne). 2024 Oct 21;11:1455715. doi: 10.3389/fmed.2024.1455715, PMID 39497852.
Xue Q, Wang N, Xue X, Wang J. Endobronchial tuberculosis: an overview. Eur J Clin Microbiol Infect Dis. 2011 Sep;30(9):1039-44. doi: 10.1007/s10096-011-1205-2, PMID 21499709.
Bai W, Ameyaw EK. Global regional and national trends in tuberculosis incidence and main risk factors: a study using data from 2000 to 2021. BMC Public Health. 2024 Jan 2;24(1):12. doi: 10.1186/s12889-023-17495-6, PMID 38166735.
Ignatius EH, Dooley KE. New drugs for the treatment of tuberculosis. Clin Chest Med. 2019;40(4):811-27. doi: 10.1016/j.ccm.2019.08.001, PMID 31731986.
Goldman AL, Braman SS. Isoniazid: a review with emphasis on adverse effects. Chest. 1972;62(1):71-7. doi: 10.1378/chest.62.1.71, PMID 4339326.
Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin myricetin and staurosporine. Mol Cell. 2000;6(4):909-19. doi: 10.1016/s1097-2765(05)00089-4, PMID 11090628.
Baqi Ma, Jayanthi K, Rajeshkumar R. Molecular docking insights into probiotics sakacin p and sakacin a as potential inhibitors of the cox-2 pathway for colon cancer therapy. Int J App Pharm. 2025;17(1):153–60. doi: 10.22159/ijap.2025v17i1.52476.
Pant K, Karpel RL, Rouzina I, Williams MC. Mechanical measurement of single-molecule binding rates: kinetics of DNA helix destabilization by T4 gene 32 protein. J Mol Biol. 2004;336(4):851-70. doi: 10.1016/j.jmb.2003.12.025, PMID 15095865.
Sherman W, Beard HS, Farid R. Use of an induced fit receptor structure in virtual screening. Chem Biol Drug Des. 2006 Jan;67(1):83-4. doi: 10.1111/j.1747-0285.2005.00327.x, PMID 16492153.
Imam SS, Imam ST, Mdwasifathar K, Kumar R, Ammar MY. Interaction between ace 2 and SARS-CoV-2, and use of EGCG and theaflavin to treat COVID-19 in initial phases. Int J Curr Pharm Sci. 2022;14(2):5-10. doi: 10.22159/ijcpr.2022v14i2.1945.
Badavath VN, Sinha BN, Jayaprakash V. Design in silico docking and predictive ADME properties of novel pyrazoline derivatives with selective human MAO inhibitory activity. Int J Pharm Pharm Sci. 2015;7(12):277-82.
Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT. Glide: a new approach for rapid accurate docking and scoring. 1. method and assessment of docking accuracy. J Med Chem. 2004 Mar 1;47(7):1739-49. doi: 10.1021/jm0306430, PMID 15027865.
Dhawale S, Gawale S, Jadhav A, Gethe K, Raut P, Hiwarale N. In silico approach targeting polyphenol as fabh inhibitor in bacterial infection. Int J Pharm Pharm Sci. 2022;14(11):25-30. doi: 10.22159/ijpps.2022v14i11.45816.
Divyashri G, Krishna Murthy TP, Sundareshan S, Kamath P, Murahari M, Saraswathy GR. In silico approach towards the identification of potential inhibitors from Curcuma amada Roxb against H. pylori: ADMET screening and molecular docking studies. Bioimpacts. 2021;11(2):119-27. doi: 10.34172/bi.2021.19, PMID 33842282.
Mulakala C, Viswanadhan VN. Could MM-GBSA be accurate enough for calculation of absolute protein/ligand binding free energies? J Mol Graph Model. 2013;46:41-51. doi: 10.1016/j.jmgm.2013.09.005, PMID 24121518.
Schroeder EK, De Souza N, Santos DS, Blanchard JS, Basso LA. Drugs that inhibit mycolic acid biosynthesis in Mycobacterium tuberculosis. Curr Pharm Biotechnol. 2002;3(3):197-225. doi: 10.2174/1389201023378328, PMID 12164478.
Kumari M, Tiwari N, Chandra S, Subbarao N. Comparative analysis of machine learning based QSAR models and molecular docking studies to screen potential anti-tubercular inhibitors against InhA of mycobacterium tuberculosis. IJCBDD. 2018;11(3):209. doi: 10.1504/IJCBDD.2018.094630.
Dasmahapatra U, Kumar CK, Das S, Subramanian PT, Murali P, Isaac AE. In-silico molecular modelling MM/GBSA binding free energy and molecular dynamics simulation study of novel pyrido-fused imidazo[4,5-c]quinolines as potential anti-tumor agents. Front Chem. 2022;10:991369. doi: 10.3389/fchem.2022.991369, PMID 36247684.
Campanico A, Moreira R, Lopes F. Drug discovery in tuberculosis new drug targets and antimycobacterial agents. Eur J Med Chem. 2018;150:525-45. doi: 10.1016/j.ejmech.2018.03.020, PMID 29549838.
Lienhardt C, Raviglione M, Spigelman M, Hafner R, Jaramillo E, Hoelscher M. New drugs for the treatment of tuberculosis: needs challenges, promise and prospects for the future. J Infect Dis. 2012;205 Suppl 2:S241-9. doi: 10.1093/infdis/jis034, PMID 22448022.