
Int J App Pharm, Vol 17, Issue 5, 2025, 203-213Original Article
MILTEFOSINE LOADED MESOPOROUS ZINC OXIDE NANOPARTICLES TO ENHANCE ORAL BIOAVAILABILITY AND SAFETY PROFILE: A PRETOXICOLOGICAL AND PHARMACOKINETIC STUDY
PARAG GHOSH1, SUBAS CHANDRA DINDA1*, DEBAJIT DEWAN2, SUKANTA ROY1, DIBYA DAS3, SOURAV DAS1, ANIRBANDEEP BOSE4*
1School of Pharmacy, the Neotia University, West Bengal, India. 2Bharat Technology, Bahirtafa, Beltala, Uluberia, Howrah, West Bengal, India. 3Department of Pharmaceutical Technology, JIS University, West Bengal, India. 4Department of Pharmaceutical Technology, Adamas University, West Bengal, India
*Corresponding author: Subas Chandra Dinda; *Email: subaschandra.dinda@tnu.in
Received: 21 Apr 2025, Revised and Accepted: 11 Jun 2025
ABSTRACT
Objective: Miltefosine, the only approved oral therapy for leishmaniasis, is clinically limited by its poor bioavailability and significant gastrointestinal and systemic toxicities. This study aimed to develop and evaluate a novel oral formulation of miltefosine encapsulated in mesoporous zinc oxide nanoparticles (MF-ZnO NPs) to enhance its pharmacokinetic Safety Profile.
Methods: MF-ZnO NPs were synthesized via a templated method, achieving a particle size of ~178 nm and a drug loading efficiency of 60%. Stability studies were conducted in simulated gastric and intestinal fluids to assess nanoparticle integrity. In vitro drug release was evaluated in PBS (pH 6.8), showing a biphasic pattern with sustained release. A 28 d subacute oral toxicity study was performed in wistar rats across escalating dose groups, with raw miltefosine as a comparator. Parameters assessed included behavioral toxicity, biochemical markers (ALT, AST, creatinine, urea, bilirubin), organ weights, and histopathological scoring. Pharmacokinetic studies in rats determined systemic exposure and bioavailability enhancements. In vivo skin deposition was also assessed after 24 h.
Results: MF-ZnO NPs demonstrated excellent physicochemical stability and controlled biphasic drug release with 95% cumulative release at 72 h. Toxicological evaluation revealed minimal hepatic and renal toxicity at low and medium doses, whereas high-dose MF-ZnO NPs and raw miltefosine induced significant organ damage. Pharmacokinetic analysis showed improved oral bioavailability (AUC₀–inf: 7375.9±609.56 vs. 5539.7±240.99 h·µg/ml) and prolonged half-life (73.84±11.32 h vs. 57.29±5.61 h) for MF-ZnO NPs. Skin deposition studies confirmed a fourfold increase in localized drug accumulation with the nanoparticle formulation.
Conclusion: Encapsulation of miltefosine in mesoporous ZnO nanoparticles significantly enhanced its oral bioavailability, reduced systemic toxicity, and improved dermal deposition. MF-ZnO NPs present a promising oral delivery strategy for safer and more effective miltefosine therapy.
Keywords: Miltefosine, Mesoporous ZnO nanoparticles, Oral bioavailability, Pharmacokinetics, Toxicity, Skin deposition, Nanomedicine, Leishmaniasis
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i5.54732 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Leishmaniasis represents one of the most significant neglected tropical diseases, with an estimated 700,000 to 1 million new cases and 20,000 to 30,000 deaths occurring annually worldwide, according to the World Health Organization (WHO). It is caused by protozoan parasites of the genus Leishmania and is transmitted through the bite of infected female phlebotomine sandflies. The disease manifests in several clinical forms, ranging from the life-threatening visceral leishmaniasis (VL), also known as kala-azar to the more disfiguring yet non-fatal cutaneous and mucocutaneous forms. VL is endemic in over 80 countries, disproportionately affecting impoverished and rural populations in South Asia, East Africa, and South America. Post-kala-azar dermal leishmaniasis (PKDL), a dermatological sequel of VL, further complicates disease management due to its chronicity, diagnostic challenges, and potential role in parasite transmission. Despite public health efforts, treatment remains limited by drug toxicity, poor patient compliance, and increasing drug resistance [1].
Among the pharmacotherapeutic options available, miltefosine is the only oral drug approved for treating VL and PKDL Its ease of administration makes it a valuable tool in disease-endemic areas with limited healthcare infrastructure. However, its clinical use is increasingly restricted by low aqueous solubility, variable absorption, long half-life, and pronounced gastrointestinal and systemic toxicities. These pharmacokinetic drawbacks necessitate high and prolonged dosing regimens, which in turn elevate the risk of adverse events and foster the emergence of drug-resistant strains of Leishmania. Moreover, concerns about teratogenicity and hepatotoxicity further limit its use in special populations, including women of childbearing age and individuals with comorbid conditions [2, 3].
To address these challenges, significant attention has turned toward nanotechnology-based drug delivery systems, which offer the ability to overcome the limitations of conventional formulations [4]. Mesoporous zinc oxide nanoparticles (ZnO NPs) have emerged as a particularly attractive candidate due to their high surface area, controllable pore structure, and biocompatibility. These particles are capable of improving oral bioavailability, offering sustained drug release, and minimizing exposure of the active drug to off-target tissues [5, 6]. Furthermore, ZnO NPs possess inherent antimicrobial properties, making them especially suitable for infectious disease contexts [7-9]. Encapsulating miltefosine in such a nanocarrier system holds the potential to optimize its pharmacokinetic profile, reduce systemic toxicity, and enhance targeted deposition notably in dermal tissues affected by PKDL.
Despite their promise, no studies have comprehensively evaluated the toxicological and pharmacokinetic implications of delivering miltefosine via mesoporous ZnO nanoparticles. A rigorous preclinical assessment encompassing behavioral toxicity, biochemical markers, organ histopathology, and systemic pharmacokinetics-is essential to determine the viability of such a formulation. This study is thus framed within the broader objective of redefining miltefosine delivery using advanced nanomedicine strategies, with the aim of improving its safety, therapeutic efficacy, and patient compliance [10-14].
By integrating principles of formulation science, toxicology, and infectious disease pharmacology, this investigation seeks to establish a rational foundation for the next generation of oral miltefosine therapies, addressing both the clinical unmet need in leishmaniasis treatment and the broader challenge of safe, effective delivery of bioactive compounds with narrow therapeutic indices.
MATERIALS AND METHODS
Materials
Mesoporous zinc oxide (ZnO) nanoparticles were synthesized in the laboratory of the School of Pharmacy, The Neotia University. The nanoparticles were prepared with a size range of 150–220 nm with an average diameter of 178.4 nm. Miltefosine, a commercially available anti-leishmanial drug, was procured from Merck, USA. The drug loading capacity of the nanoparticles was optimized to approximately 60%. Prior to initiating the study, ethical approval was obtained from the Institutional Animal Ethics Committee (IAEC) under the protocol number TOXSC (R) MSPNP 0123 and PKPD (R) MSPNP 0123 from IAEC Registration No. 1938/PO/Rc/S/17/CPCSEA, in compliance with the CPCSEA guidelines.
Drug loading efficiency determination
Miltefosine loading in mesoporous ZnO nanoparticles was quantified using UV–Visible spectrophotometry. A known quantity of MF-ZnO NPs was dispersed in methanol and subjected to mild sonication to extract the encapsulated drug. The solution was filtered and analyzed at 269 nm using a UV–Vis spectrophotometer. A calibration curve of standard miltefosine solutions was used for quantification. Drug loading efficiency (%) was calculated as:
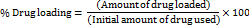
Stability study in simulated gastric and intestinal fluids
The stability of MF-ZnO-NPs was evaluated in simulated gastric fluid (SGF; pH 1.2) and simulated intestinal fluid (SIF; pH 6.8) to mimic gastrointestinal conditions. SGF was prepared according to USP guidelines by dissolving 0.2% NaCl and adjusting the pH to 1.2 using concentrated HCl. SIF was prepared using 0.68% monobasic potassium phosphate and adjusting the pH to 6.8 with NaOH. MF-ZnO-NPs were dispersed in each medium at a concentration of 1 mg/ml and incubated at 37 °C under gentle shaking. Particle size distribution and polydispersity index (PDI) were measured at 0, 2, and 4 h using Dynamic Light Scattering (DLS), while zeta potential was assessed using a Zetasizer Nano ZS (Malvern Instruments, UK). Samples were visually inspected for any signs of aggregation, and drug retention was evaluated by centrifuging the suspension at 15,000 rpm for 30 min followed by quantification of miltefosine in the supernatant using UV-visible spectroscopy (Shimadzu UV-1800) after treated with ammonium ferrithiocyanate (NH4Fe[NCS]4) dye at 460 nm.
In vitro drug release profile
The in vitro release behavior of miltefosine from MF-ZnO-NPs was studied in phosphate-buffered saline (PBS; pH 6.8) to simulate intestinal conditions. A known quantity of MF-ZnO-NPs equivalent to 10 mg of miltefosine was placed in a dialysis bag (MWCO 12,000 Da) and immersed in 100 ml of PBS maintained at 37±0.5 °C under continuous stirring at 100 rpm. At predetermined time points (0, 1, 2, 4, 6, 12, 24, 36, 48, 60, and 72 h), 2 ml aliquots were withdrawn and replaced with an equal volume of fresh medium. The amount of released drug was measured using UV–visible spectroscopy was performed using a Shimadzu UV-1800 spectrophotometer at 460 nm, following treatment with ammonium ferrithiocyanate (NH₄Fe[NCS]₄) dye and appropriate sample dilution. All experiments were performed in triplicate. The release kinetics were analyzed and fitted to various models (zero-order, first-order, Higuchi, and Korsmeyer-Peppas) to determine the drug release mechanism.
Pharmacological evaluation
Experimental animals
Adult male and female wistar rats (8–10 w old, weighing 200–230 g) were procured and housed under standard laboratory conditions (12 h light/dark cycle, 22±2 °C, 50±10% relative humidity) with ad libitum access to food and water. The study was conducted following the OECD Guideline and approved by the Institutional Animal Ethics Committee (IAEC) [15].
Study design
The experimental rats were randomly allocated into four distinct groups, each comprising six animals (n=6 per group). The group size aligns with OECD 407 and 425 guidelines for subacute toxicity testing, balancing scientific validity and ethical considerations. Group 1 served as the control and was administered the vehicle alone, specifically phosphate-buffered saline (PBS). The remaining three groups received increasing oral doses of miltefosine-loaded mesoporous ZnO nanoparticles (MF-ZnO-NPs): Group 2 (Low Dose) received 50 mg/kg, Group 3 (Medium Dose) received 100 mg/kg, Group 4 (High Dose) received 200 mg/kg of MF-ZnO-NPs and an additional Group 5 was administered miltefosine at a dose of 100 mg/kg body weight, matching the medium dose used in the MF-ZnO-NPs nanoparticle-treated group. This parallel group served as a critical comparator for evaluating the safety advantage, if any, conferred by the nanoparticle-based drug delivery system. All treatments were given orally once daily for a continuous period of 28 days. All animals were housed under standard laboratory conditions with free access to food and water. Clinical observations were recorded daily to detect any signs of systemic toxicity or behavioral changes. Body weight and food intake were monitored at regular intervals throughout the study period. At the end of the 28 d treatment regimen, animals were euthanized by the end of the study period, animals were euthanized using CO₂ inhalation in a controlled chamber, following humane endpoints, and blood samples were collected for serum biochemical analysis (ALT, AST, creatinine, urea, and total bilirubin). Organs (Stomach, liver, lungs, Heart and kidney) were excised and processed for histopathological examination. Slides were stained with hematoxylin and eosin (H and E) and examined under a microscope. Histopathological analysis of liver and kidney sections was performed to assess structural alterations induced by the treatment. In addition to qualitative assessment, a semi-quantitative scoring system (0–3 scale) was used to evaluate the degree of hepatic necrosis, hepatic inflammation, renal tubular degeneration, and renal inflammation [16, 17].
In vivo pharmacokinetic study
Experimental design
Wistar rats, comprising six animals per group with an equal gender distribution of three males and three females, were utilized in the pharmacokinetic evaluation. Prior to dosing, the rats were subjected to overnight fasting to standardize gastrointestinal conditions and minimize variability in absorption profiles. The animals were subsequently allocated into two distinct groups. Group A received an oral administration of free miltefosine at a dose of 50 mg/kg body weight. In contrast, Group B was administered miltefosine-loaded mesoporous zinc oxide nanoparticles (MF-ZnO-NPs) at a dosage equivalent to 50 mg/kg of miltefosine, also via the oral route. This comparative approach was designed to investigate and elucidate the impact of nanoparticle encapsulation on the oral bioavailability, pharmacokinetics, and potential safety advantages relative to the conventional free drug formulation. Blood samples, each approximately 200 µl in volume, were systematically collected from the rats at predetermined time intervals of 0 (immediately before dosing), 0.5, 1, 2, 3, 6, 8, 12, 24, 48, 72, 96, and 120 h following oral administration. The sampling was carefully performed via retro-orbital puncture, a technique chosen for its minimal invasiveness and effectiveness in ensuring sufficient sample volume performed under light isoflurane anesthesia (2–3% in oxygen via a nose cone) to minimize pain and distress. Immediately following collection, blood samples were centrifuged at 3,000 RPM for 10 min to efficiently separate the plasma fraction. The resultant plasma was then carefully isolated, aliquot, and stored at a temperature of-80 °C, ensuring the preservation of analyte stability until subsequent pharmacokinetic analyses were conducted. This rigorous sampling and storage protocol was crucial to reliably measure the concentration-time profiles and accurately characterize the pharmacokinetic behavior of both free miltefosine and MF-ZnO-NPs nanoparticle formulations [18].
In vivo skin deposition study (24 h analysis)
Post-kala-azar dermal leishmaniasis (PKDL) develops as a cutaneous manifestation following an episode of visceral leishmaniasis (VL). Existing therapies for PKDL remain suboptimal, and limited information is available on the localization of antileishmanial agents within the skin [19]. This study was conducted to assess and compare the skin deposition profile of miltefosine when administered orally as miltefosine-loaded ZnO nanoparticles (MF-ZnO-NPs) versus raw miltefosine over a 24 h period [20]. For this purpose, six healthy Wistar rats, each weighing between 250 and 300 gs, were selected and randomly divided into two groups, ensuring an equitable distribution for comparative analysis. Group A was orally administered MF-ZnO-NPs at a dose equivalent to 10 mg/kg of miltefosine, while Group B received an oral dose of raw miltefosine at 10 mg/kg. Administration was precisely performed using oral gavage to ensure accurate dosing and reproducibility. At 24 h’ post-dose, skin tissue samples were carefully excised from the dorsal region of each animal. These collected tissues were then homogenized thoroughly and analyzed for miltefosine content utilizing a validated LC-MS/MS method to ensure sensitivity, specificity, and accuracy in quantification.
Results obtained were statistically expressed as mean values±standard deviation (SD), calculated based on three independent readings per rat (n = 6). The experimental protocols were strictly conducted in compliance with established ethical standards and guidelines, and prior approval was obtained from the Institutional Animal Ethics Committee (IAEC) Registration No. 1938/PO/Rc/S/17/CPCSEA, approval no PKPD (R) MSPNP 0123 in compliance with the CPCSEA guidelines.
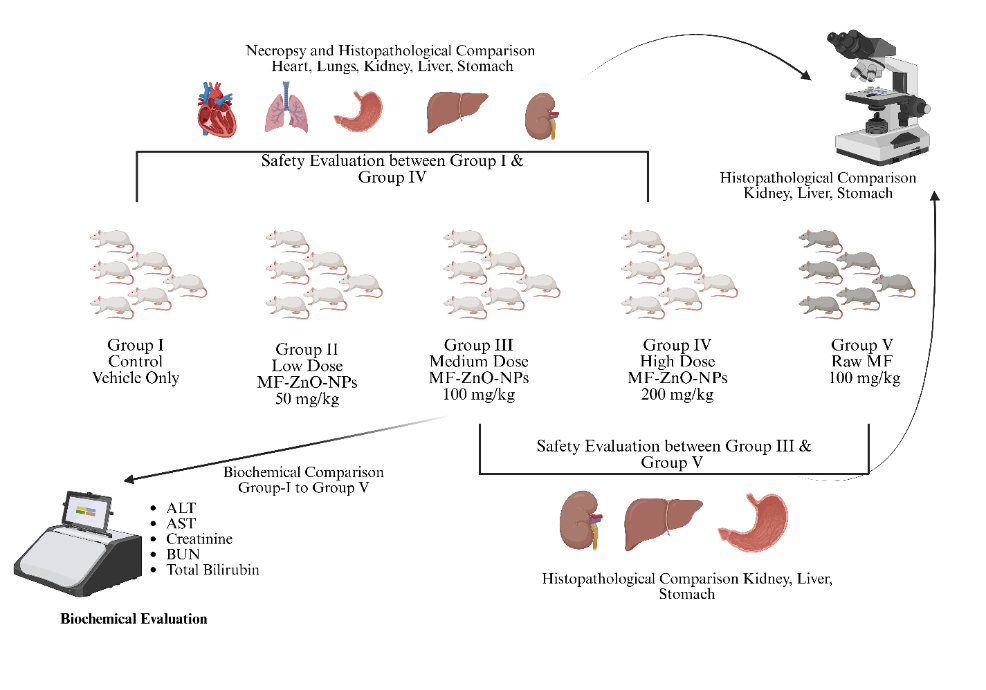
Fig. 1: The experimental design involves a comparative toxicity evaluation of miltefosine-loaded zinc oxide nanoparticles (MF-ZnO-NPs) in wistar rats. Five groups were studied: Group I received only the vehicle (control), while Groups II, III, and IV received increasing oral doses of MF-ZnO-NPs at 50 mg/kg (low dose), 100 mg/kg (medium dose), and 200 mg/kg (high dose), respectively. Group V received raw miltefosine at a dose of 100 mg/kg to serve as a comparator for unformulated drug toxicity. Biochemical evaluations were conducted for Groups I to V to assess liver and kidney function using ALT, AST, creatinine, urea (BUN), and total bilirubin levels. Safety assessments were made between Group I and Group IV to evaluate the effects of the highest nanoparticle dose, and between Group III and Group V to compare the safety of the nanoparticle formulation with raw miltefosine. Histopathological examinations were performed post-necropsy. Group I and IV were compared across the heart, lungs, liver, kidney, and stomach, whereas Group III and V were compared with focus on kidney, liver, and stomach to determine structural and cellular alterations induced by the treatments. Publication License available on: https://biorender.com/gq3c44h
Drug quantification
The plasma concentrations of miltefosine were quantified employing a robust and highly sensitive LC-MS/MS analytical method, utilizing the API 4000 triple quadrupole mass spectrometer from AB Sciex. Chromatographic separation was effectively achieved using a Phenomenex Kinetex C18 analytical column (50×3 mm, 5 µm particle size). The analysis was performed under isocratic elution conditions, employing a carefully optimized binary mobile phase composed of a 0.069% ammonia solution mixed with 30 mmol ammonium acetate buffer adjusted to pH 8.90, and 10 mmol ammonia dissolved in methanol. The mobile phase was maintained at a consistent flow rate of 0.6 ml/min, allowing a short and efficient chromatographic run of 6 min. Plasma samples were meticulously prepared by employing a protein precipitation technique using ice-cold acetonitrile, facilitating rapid sample processing and effective removal of interfering plasma proteins. Calibration standards prepared in plasma ranged from 2 ng/ml to 2000 ng/ml and exhibited excellent linearity, with correlation coefficients (R²) consistently equal to 0.999. For quantification, miltefosine and the internal standard metoprolol were monitored by multiple reaction monitoring (MRM) transitions at mass-to-charge ratios (m/z) of 408.1→124.9 and 268.2→116.2, respectively, under positive electrospray ionization mode. Critical instrumental parameters, such as curtain gas (maintained at 10 psi), collision energy (optimized at 45 eV for miltefosine), and interface heater settings, were systematically adjusted and optimized to achieve high sensitivity, selectivity, and reproducibility of the analytical assay. This rigorous analytical approach ensured accurate, precise, and reliable quantification of miltefosine concentrations in plasma samples collected throughout the pharmacokinetic study [18, 21].
Statistical analysis
All experimental results were expressed as mean±standard deviation (SD), and each group consisted of six animals (n = 6). The biochemical and behavioral parameters measured across multiple time points were analyzed using one-way repeated measures ANOVA, followed by Bonferroni-adjusted post hoc comparisons to account for multiple testing. Prior to ANOVA, the assumptions of normality and homogeneity of variances were assessed using the Shapiro–Wilk test and Levene’s test, respectively. No violations of these assumptions were observed (p>0.05 for all comparisons). Statistical analyses were conducted using GraphPad Prism (v9.0), and significance was set at p<0.05.
RESULTS
Stability study in SGF and SIF
MF-ZnO-NPs exhibited excellent stability in both SGF and SIF over the observation period. As shown in table 1, the initial particle size was 178.2±5.4 nm with a PDI of 0.214±0.01, and no significant change was observed after 4 h of incubation in either medium (183.4±7.1 nm in SGF and 181.2±6.5 nm in SIF). The zeta potential remained stable around-22 to-24 mV, indicating sufficient colloidal stability to prevent aggregation. No visible agglomeration was observed, and the drug retention remained above 75% in both media. These findings confirm the physicochemical stability of MF-ZnO-NPs under gastrointestinal conditions, ensuring that the nanoparticles can maintain their integrity during oral transit, a critical attribute for effective drug delivery.
Table 1: Stability of MF-ZnO-NPs in simulated gastric fluid (SGF) and simulated intestinal fluid (SIF)
| Parameter | SGF (pH 1.2) | SIF (pH 6.8) |
| Initial particle size (nm) | 178.2±5.4 | 185.2±3.4 |
| Polydispersity Index (PDI) | 0.214±0.01 | 0.234±0.02 |
| Zeta Potential (mV) | -22.5±1.3 | -23.8±1.4 |
| Size after 2 h (nm) | 180.9±6.2 | 179.5±5.8 |
| Size after 4 h (nm) | 183.4±7.1 | 181.2±6.5 |
| Aggregation Observed (Y/N) | No | No |
| Drug Retention (%) after 4 h | 81.2±2.1 | 76.2±2.3 |
Data is given in mean±SD, n=3
In vitro drug release profile
The drug release study demonstrated a biphasic release pattern, characteristic of mesoporous nanoparticulate systems. An initial burst release of approximately 28% within the first 6 h was observed, which is likely attributable to the desorption of miltefosine molecules loosely bound to the nanoparticle surface. This was followed by a sustained release phase, with cumulative drug release reaching 98% at 60 h (table 2). Kinetic modeling revealed that the release was best described by first-order kinetics (R² = 0.9907), indicating concentration-dependent drug release, while the Higuchi model (R² = 0.9886) confirmed diffusion through the porous ZnO matrix as the predominant mechanism. Zero-order fitting (R² = 0.9768) suggested a near constant release component, desirable for sustained action, whereas the Korsmeyer–Peppas model (R² = 0.9755) indicated anomalous transport involving both diffusion and matrix governed mechanism.. Such a release profile is highly desirable for oral formulations, ensuring both an early therapeutic effect and prolonged drug availability in systemic circulation.
Table 2: In vitro drug release profile of MF-ZnO-NPs
| Time (h) | Cumulative drug release (%) |
| 0 | 0 |
| 1 | 10±1.2 |
| 2 | 16±1.4 |
| 4 | 22±1.5 |
| 6 | 28±1.7 (Initial Burst) |
| 12 | 58±2.0 |
| 24 | 72±2.2 |
| 36 | 83±2.5 |
| 48 | 91±2.4 |
| 60 | 98±2.1 |
| 72 | 98±2.9 (Sustained Phase) |
Data is given in mean±SD, n=3
The in vitro drug release behavior of miltefosine from MF-ZnO-NPs was evaluated in phosphate-buffered saline (PBS, pH 6.8) at 37 °C to mimic intestinal conditions. The release profile exhibited a biphasic pattern, characterized by an initial burst phase followed by sustained release. During the initial phase, a cumulative release of 28±1.7% was observed within the first 6 h, indicating the rapid release of surface-adsorbed drug molecules. This controlled burst is beneficial for achieving early therapeutic plasma concentrations post oral administration. Subsequently, the formulation exhibited a sustained release phase, with cumulative drug release reaching 58±2.0% at 12 h, 72±2.2% at 24 h, and 83±2.5% at 36 h. The release continued progressively, reaching 91±2.4% at 48 h and 98±2.1% at 60 h, with a plateau observed at 98±2.9% by 72 h, indicating near-complete drug release. The overall release kinetics were best described by the Higuchi model (R²>0.98), suggesting that the drug release from MF-ZnO-NPs was predominantly governed by a diffusion-controlled mechanism through the mesoporous matrix. This biphasic release behavior, combining an initial burst with sustained delivery, highlights the potential of MF-ZnO-NPs as an effective oral delivery system capable of maintaining prolonged therapeutic drug levels.
Pharmacological evaluation study
Behavioral toxicity evaluation based on food consumption and body weight
The assessment of food intake and body weight served as vital behavioral indicators of systemic health during the 28 d subacute toxicity study shown in table 3. Animals in the control group (Group I) exhibited normal feeding behavior and body weight (13.82±4.23 g/day and 259.3±10.1 g, respectively), suggesting stable physiological status under standard conditions. In contrast, animals receiving low and medium doses of MF-ZnO-NPs (Groups II and III) showed mild, non-significant reductions in food intake and body weight, indicating good tolerability at these doses. However, a significant decline in both parameters was observed in the high-dose MF-ZnO-NPs group (Group IV), with food intake reduced to 5.34±1.4 g/day (***p<0.001) and body weight to 249.8±7.4 g (**p<0.01), suggesting moderate systemic stress. The most pronounced effect was noted in the raw miltefosine-treated group (Group V), which showed the lowest food intake (4.92±1.1 g/day; ***p<0.001) and significant weight loss (235.5±7.1 g; ***p<0.001), reflecting marked intolerance likely due to gastrointestinal or systemic toxicity. These findings demonstrate a clear dose-and formulation-dependent difference in tolerability, with MF-ZnO-NPs offering a comparatively safer profile than raw miltefosine, particularly at therapeutic dose levels.
Table 3: Summary of behavioral parameters on day 28
| Parameters | Group I (control) | Group II (Low dose) | Group III (Medium dose) | Group IV (High dose) | Group V (Raw miltefosine) |
| Food Intake (g/day) | 13.82±4.23 | 11.67±4.5 | 10.56±4.2 | 5.34±1.4*** | 4.92±1.1*** |
| Body Weight (g) | 259.3±10.1 | 262.2±11.5 | 256.0±4.4 | 249.8±7.4** | 235.5±7.1*** |
**p<0.01, ***p<0.001 vs. Group I (Control) using bonferroni-adjusted post hoc test (n=6 each group)
Biochemical evaluation following 28 d oral administration of MF-ZnO-NPs nanoparticles
Biochemical evaluation of hepatic and renal function markers revealed a distinct dose-dependent toxicity pattern across treatment groups, shown in table 4. In the control group (Group I), all parameters remained within normal physiological limits, serving as a baseline reference. Group II (Low Dose MF-ZnO-NPs) showed mild but statistically significant elevations in serum ALT (37.39±0.19 U/l; *p<0.05), AST (168.65±0.22 U/l; *p<0.05), creatinine (0.55±0.03 mg/dl; *p<0.05), and total bilirubin (0.29±0.02 mg/dl; *p<0.05), while BUN (37.74±0.45 mg/dl) remained statistically non-significant, suggesting early hepatic and renal adaptive responses without marked toxicity. In Group III (Medium Dose MF-ZnO-NPs), the elevations became more prominent, with ALT (44.26±0.21 U/l; **p<0.01), AST (205.73±0.26 U/l; **p<0.01), creatinine (0.71±0.04 mg/dl; **p<0.01), and total bilirubin (0.37±0.03 mg/dl; **p<0.01) all showing statistically significant changes, along with a moderate rise in BUN (47.03±0.51 mg/dl; **p<0.01), indicating moderate hepatocellular and renal compromise. In Group IV (High Dose MF-ZnO-NPs), these alterations were substantial and highly significant: ALT (51.66±0.32 U/l; ***p<0.001), AST (263.60±0.41 U/l; ***p<0.001), creatinine (1.34±0.06 mg/dl; ***p<0.001), total bilirubin (1.09±0.06 mg/dl; ***p<0.001), and BUN (61.09±0.57 mg/dl; ***p<0.001), indicating advanced hepatic injury and renal dysfunction. The most severe alterations were observed in Group V (Raw Miltefosine), which recorded the highest values across all parameters ALT (57.10±0.27 U/l), AST (273.53±0.36 U/l), creatinine (1.23±0.05 mg/dl), total bilirubin (1.11±0.05 mg/dl), and BUN (60.90±0.59 mg/dl) each with ***p<0.001 vs. control, reflecting profound hepatic and renal toxicity. These results confirm that while both high-dose MF-ZnO-NPs and raw miltefosine induce biochemical evidence of organ damage, the nanoparticle formulation demonstrates a relatively safer profile at low and medium doses, thereby underscoring its potential as a more tolerable oral delivery system for miltefosine.
Table 4: Summary of biochemical parameters on Day 28 with statistical significance
| Biochemical parameters | Group I (Control) | Group II (Low dose) | Group III (Medium dose) | Group IV (High dose) | Group V (Raw miltefosine) |
| ALT (U/l) | 33.12±0.21 | 37.39±0.19* | 44.26±0.21** | 51.66±0.32*** | 57.10±0.27*** |
| AST (U/l) | 156.15±0.38 | 168.65±0.22* | 205.73±0.26** | 263.60±0.41*** | 273.53±0.36*** |
| Creatinine (mg/dl) | 0.31±0.01 | 0.55±0.03* | 0.71±0.04** | 1.34±0.06*** | 1.23±0.05*** |
| Total Bilirubin (TBIL) (mg/dl) | 0.17±0.01 | 0.29±0.02* | 0.37±0.03** | 1.09±0.06*** | 1.11±0.05*** |
| Blood Urea Nitrogen (BUN) (mg/dl) | 34.13±0.43 | 37.74±0.45 | 47.03±0.51** | 61.09±0.57*** | 60.90±0.59*** |
*p<0.05, **p<0.01, ***p<0.001 vs. Group I (Control) using Bonferroni-adjusted post hoc test. (n=6 each group)
Overall, the data underscore a dose-dependent hepatic and renal burden shown in fig. 2, with the MF-ZnO-NPs nanoparticle formulation showing improved biochemical tolerance at lower doses compared to the raw drug, which elicited pronounced toxicity. These findings reinforce the protective role of the nanoparticle delivery system in mitigating systemic organ damage while sustaining drug efficacy.
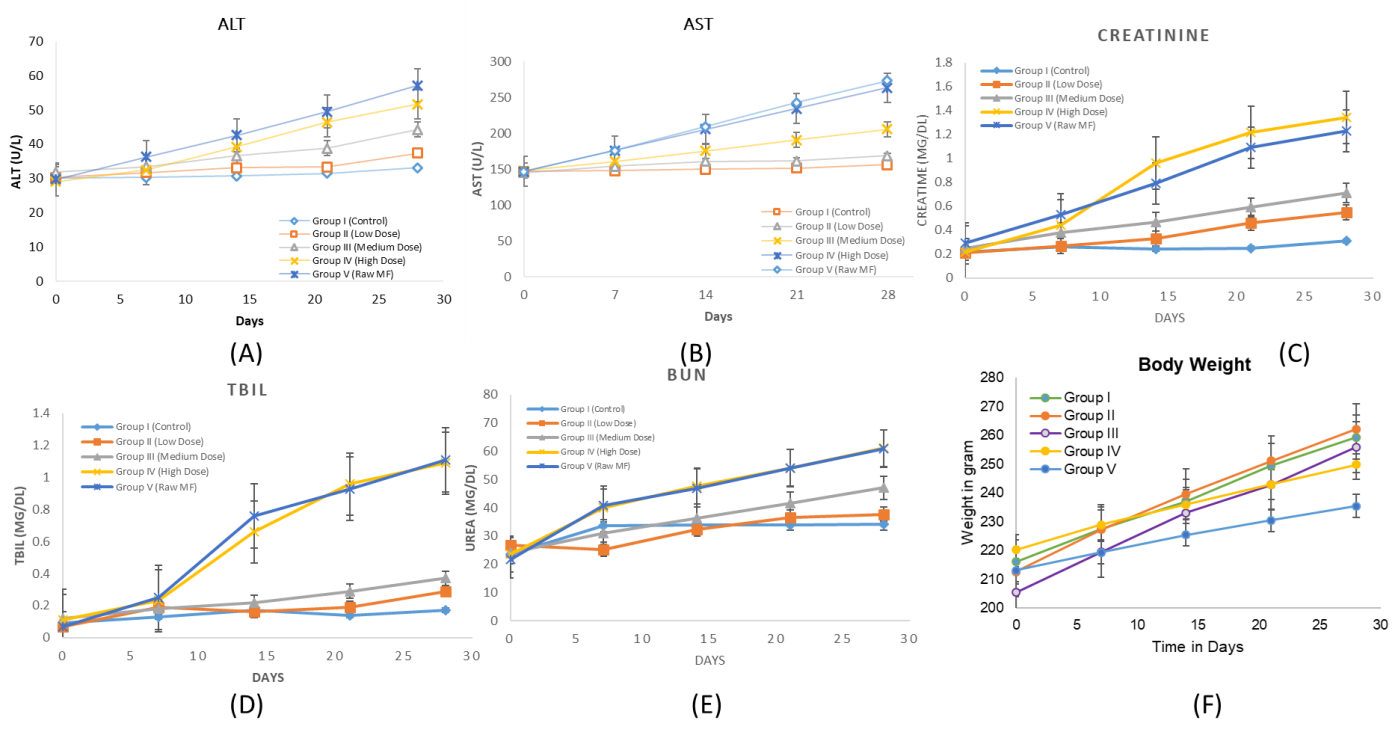
Fig. 2: Illustrates the temporal progression of serum biochemical and behavioral parameters over a 28 d period following oral administration of free miltefosine and miltefosine-loaded mesoporous ZnO nanoparticles (MF-ZnO-NPs NPs) in wistar rats. (A) shows ALT levels, while (B) represents AST levels, both of which increased in a dose-dependent manner, with the highest elevations observed in Group IV (High Dose MF-ZnO-NPs) and Group V (Raw Miltefosine), indicating significant hepatic stress. (C) depicts serum creatinine concentrations, which also rose notably in the high-dose and raw drug groups, suggesting impaired renal function. Similarly, (D) (total bilirubin) and (E) (BUN) further support the presence of hepatobiliary and renal toxicity at higher doses, whereas Groups II (Low Dose) and III (Medium Dose) maintained relatively stable biochemical values, indicating better systemic tolerability. Panel F presents body weight trends, where consistent weight gain was observed in Groups I to III, while Groups IV and V exhibited blunted growth trajectories, aligning with the biochemical evidence of toxicity. Collectively, these findings confirm a dose-dependent toxicity profile and highlight the improved safety of MF-ZnO-NPs formulations at lower doses. Data are presented as mean±standard deviation (n = 6 per group)
Histopathological findings
Necropsy findings
Analysis of absolute organ weights in table 5 revealed notable dose-dependent changes primarily in the liver and kidneys, which are critical target organs for miltefosine-induced toxicity. The control group (Group I) exhibited normal organ weights across all examined tissues, serving as the baseline for comparison. Groups II (Low Dose) and III (Medium Dose) MF-ZnO-NPs showed slight, non-significant increases in liver and kidney weights, suggestive of mild physiological adaptation without overt pathology. However, in Group IV (High Dose MF-ZnO-NPs), there was a statistically significant increase in liver weight (9.5±0.5 g; **p<0.01) and kidney weight (2.2±0.3 g; **p<0.01) compared to control, indicating organ hypertrophy likely associated with moderate inflammatory or degenerative changes. Group V (Raw Miltefosine) showed the highest mean liver (9.6±0.5 g) and kidney weights (2.3±0.3 g), both significantly elevated (**p<0.01), which is consistent with the observed histological evidence of hepatocellular necrosis and glomerular inflammation. In contrast, heart, stomach, and lung weights showed only marginal variations across groups, with no statistically significant differences, suggesting minimal cardiopulmonary or gastric hypertrophy at the tested doses. These findings reinforce the histopathological and biochemical evidence that MF-ZnO-NPs reduce systemic toxicity compared to raw miltefosine, particularly at low and medium doses.
Table 5: Summary of organ weights on Day 28
| Organ weight (g) | Group I (Control) | Group II (Low dose) | Group III (Medium dose) | Group IV (High dose) | Group V (Raw miltefosine) |
| Heart | 1.2±0.1 | 1.3±0.1 | 1.4±0.1 | 1.5±0.1 | 1.4±0.1 |
| Stomach | 1.5±0.1 | 1.6±0.1 | 1.7±0.1 | 1.8±0.1 | 1.7±0.1 |
| Liver | 8±0.5 | 8.5±0.5 | 9±0.5 | 9.5±0.5** | 9.6±0.5** |
| Kidney | 1.8±0.2 | 1.9±0.2 | 2.0±0.2 | 2.2±0.3** | 2.3±0.3** |
| Lungs | 1.5±0.2 | 1.6±0.2 | 1.7±0.2 | 1.8±0.3 | 1.8±0.3 |
**p<0.01 vs. Group I (Control) using Bonferroni-adjusted post hoc test (n=6 each group)
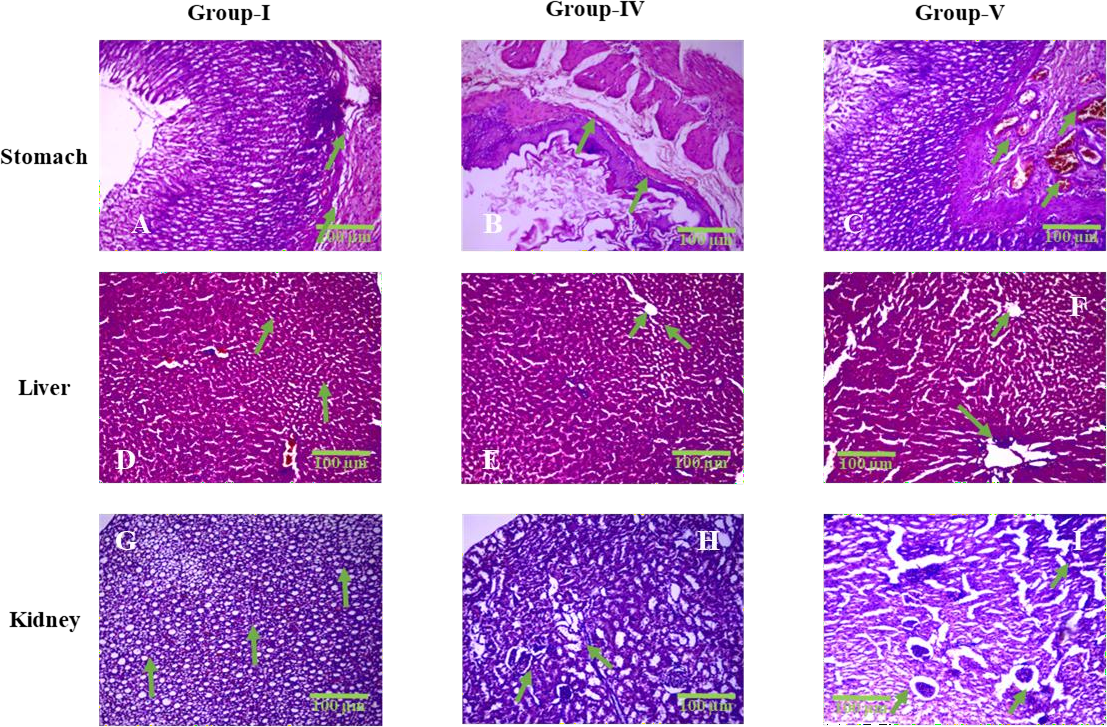
Fig. 3: Representative histopathological sections of the stomach, liver, and kidney from Group I (Control), Group IV (High Dose MF-ZnO-NPs), and Group V (Raw Miltefosine), stained with hematoxylin and eosin (HandE) and observed under 400× magnification. All images include 100 µm scale bars for reference. In the stomach sections (A–C), Group I displays intact mucosal epithelium and submucosa with no pathological changes (green arrows indicate preserved epithelial lining), whereas Group IV shows mild to moderate epithelial disruption and submucosal edema (B; green arrows mark mucosal folding and thinning), and Group V reveals severe epithelial erosion and dense inflammatory infiltration (C; green arrows indicate ulcerated regions and infiltrating leukocytes), corresponding to semi-quantitative inflammation scores of 0, 1–2, and 3, respectively. In the liver sections (D–F), Group I exhibits normal hepatic cords and central veins (green arrows in D), Group IV presents moderate hepatocellular degeneration and sinusoidal dilation (E; green arrows highlight swollen sinusoids), and Group V shows extensive centrilobular necrosis, sinusoidal congestion, and lymphocytic infiltration (F; green arrows indicate necrotic foci and disrupted hepatic cords), corresponding to necrosis scores of 0, 2, and 3. In the kidney sections (G–I), Group I demonstrates intact glomeruli and tubular structures (green arrows in G), Group IV exhibits moderate tubular degeneration and interstitial inflammation (H; green arrows highlight tubular dilation and interstitial cell infiltration), and Group V shows severe peri-glomerular inflammation, glomerular hypercellularity, and loss of tubular integrity (I; green arrows indicate pathological glomerular changes), with semi-quantitative scores ranging from 0 to 3. These histological findings visually support the graded tissue-level toxicity observed across treatment groups and reinforce the protective effect of MF-ZnO-NPs compared to raw miltefosine
Histopathological findings
Histopathological examination of gastric tissues revealed a graded pattern of mucosal response across treatment groups, summarized in table 6. Group I (Control) exhibited normal gastric histoarchitecture with an intact epithelial lining and no signs of inflammation or tissue disruption, indicating the absence of gastric toxicity (scores: epithelial disruption = 0, inflammation = 0). Group IV (High Dose MF-ZnO-NPs) showed mild toxicity characterized by subtle mucosal changes and mild epithelial irregularity, with a score of 1 for epithelial disruption and 1 for inflammation. Group V (Raw Miltefosine) displayed the most pronounced gastric alterations, including epithelial disruption (score = 2) and marked inflammatory cell infiltration (score = 3) within the mucosa, consistent with mucosal irritation and early signs of ulceration.
The histopathological evaluation of hepatic tissues revealed a progressive pattern of liver alterations correlating with dose intensity. Group I (Control) displayed completely normal hepatic architecture (scores: necrosis = 0, inflammation = 0). Group II (Low Dose MF-ZnO-NPs) and Group III (Medium Dose MF-ZnO-NPs) showed mild sinusoidal dilation and scattered hepatocellular changes (scores ranging from 0–1 and 1, respectively). Group IV (High Dose MF-ZnO-NPs) demonstrated moderate hepatocellular degeneration, sinusoidal congestion, and focal necrosis (score = 2 for both necrosis and inflammation). In contrast, Group V (Raw Miltefosine) exhibited extensive centrilobular necrosis, pronounced sinusoidal congestion, and widespread inflammatory infiltration, reflected by the highest scores (3 for both necrosis and inflammation). These findings confirm a clear dose-dependent hepatotoxic response, with the nanoparticle formulation offering relative hepatic protection compared to the free drug.
Histopathological assessment of renal tissues revealed similar dose-correlated trends. Group I (Control) showed preserved glomeruli and intact tubular structures without inflammatory changes (scores: tubular degeneration = 0, inflammation = 0). Group II and Group III displayed minor interstitial edema and mild cellular degeneration (scores 0–1 and 1–2, respectively), indicative of a mild adaptive tissue response. Group IV (High Dose MF-ZnO-NPs) exhibited moderate tubular degeneration and interstitial inflammation (scores 2–3 for degeneration, 2 for inflammation), while Group V (Raw Miltefosine) demonstrated severe peri-glomerular inflammation, glomerular hypercellularity, and pronounced tubular damage (score = 3 for both parameters), confirming significant nephrotoxic insult.
All histopathological sections were evaluated under 400× magnification using high-resolution imaging shown in fig. 3. Each fig. included 100 µm scale bars and clearly annotated histological landmarks such as hepatocellular necrosis, tubular lysis, glomerular congestion, and epithelial disruption, thereby enhancing interpretability.
Table 6: Semi-quantitative histopathological scoring on day 28 (Stomach, liver and kidney)
| Groups | Liver necrosis | Liver inflammation | Kidney tubular degeneration | Kidney inflammation | Stomach epithelial disruption | Stomach inflammation |
| Control | 0 | 0 | 0 | 0 | 0 | 0 |
| Low Dose | 0–1 | 0–1 | 0–1 | 0–1 | 0 | 0 |
| Medium Dose | 1 | 1 | 1–2 | 1 | 0–1 | 0–1 |
| High Dose | 2 | 2 | 2–3 | 2 | 1 | 1 |
| Raw Miltefosine | 3 | 3 | 3 | 3 | 2 | 3 |
Note: Scoring scale: 0 = none; 1 = mild; 2 = moderate; 3 = severe (n=6 Each Group)
In vivo pharmacokinetic study
Drug quantification
The bioanalytical method underwent thorough validation and successfully metal predefined acceptance criteria outlined by relevant regulatory guidelines, confirming its suitability and robustness for accurate miltefosine quantification. The assay exhibited excellent precision, demonstrated by intra-and inter-day coefficients of variation (CV%) consistently within the range of 2.5% to 7.5%, underscoring the method's reproducibility and reliability. Accuracy was similarly robust across the entire analytical spectrum, with measured values consistently falling between 90.9% and 109%, thereby ensuring confidence in the reported concentrations. Additionally, the method displayed superior sensitivity, established by a lower limit of detection (LLOD) of 1.35 ng/ml and a lower limit of quantification (LLOQ) at 3.81 ng/ml, enabling precise measurement of even very low concentrations of miltefosine. To further guarantee the reliability of quantification, quality control samples were meticulously prepared at low (LQC: 23.43 ng/ml), medium (MQC: 187.5 ng/ml), and high (HQC: 375.00 ng/ml) concentration levels, thereby effectively covering the entire dynamic analytical range. Stability studies further reinforced method robustness, demonstrating that miltefosine concentrations remained stable and within acceptable limits during various stress conditions, including bench-top stability, multiple freeze-thaw cycles, and short-term storage. Recovery evaluations indicated consistently high extraction efficiency, exceeding 85%, while the calculated purity of miltefosine was recorded at 100.975%, fully meeting the stringent specifications essential for reliable bioanalytical assays.
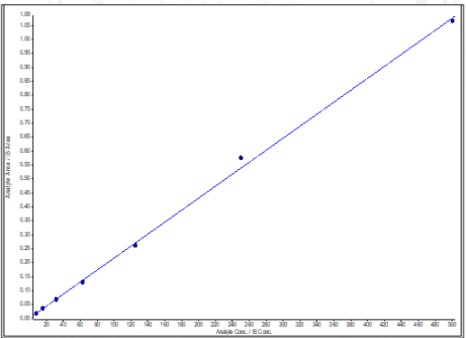
Fig. 4: Calibration curve of Miltefosine with LLOD 1.35 ng/ml; LLOQ 3.81 ng/ml; LQC 23.43 ng/ml; MQC 187.5 ng/ml; and HQC 375.00 ng/ml. (r2=0.999)
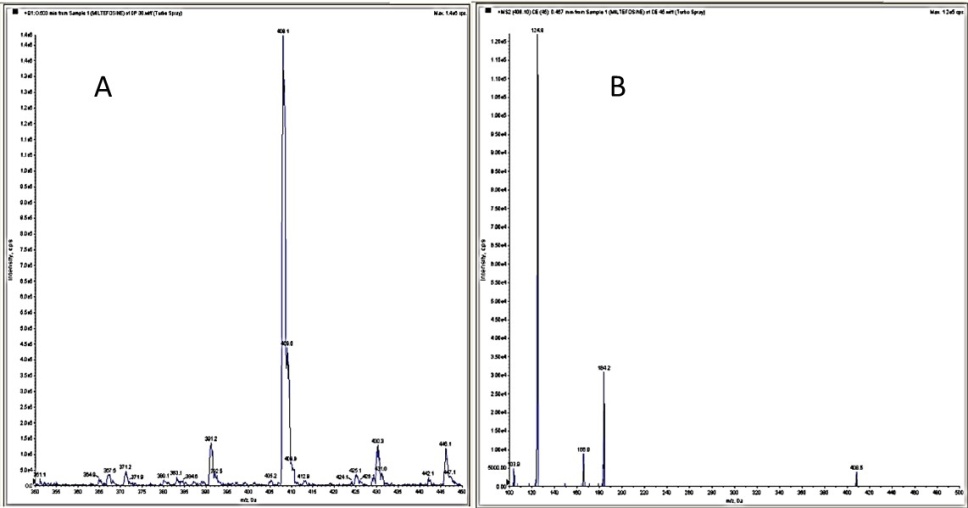
Fig. 5: (A) Miltefosine standard (Q1 scan) parent ion 408.1; (B) Miltefosine standard (MS2 scan) product ion 124.9 (Q1/Q3 transitions of 408.1/124.9 and 268.2/116.2)
This validated method provides a reliable and reproducible tool for the accurate quantification of miltefosine in plasma, suitable for clinical and pharmacokinetic applications.
Pharmacokinetic evaluation
The pharmacokinetic evaluation revealed a distinct improvement in the oral bioavailability and pharmacokinetic behavior of miltefosine when delivered via mesoporous ZnO nanoparticles (MF-ZnO-NPs) compared to the conventional free drug shown in table 7. Although both formulations exhibited the same Tmax of 24 h, indicating a similar absorption rate, significant differences were observed in the extent and duration of systemic exposure. The MF-ZnO-NPs group (Group B) achieved a higher AUC0–t (4820.5±160.5 h·µg/ml) and AUC0–inf (7375.9±609.56 h·µg/ml) compared to the free miltefosine group (Group A) with AUC0–t of 4229.4±125.14 and AUC0–inf of 5539.7±240.99, reflecting a marked enhancement in overall drug exposure due to nanoparticle encapsulation. The relative bioavailability (%RB) of the MF-ZnO-NPs formulation was calculated at 133.14%, confirming superior systemic availability compared to the raw drug. Moreover, the MF-ZnO-NPs group exhibited a reduced elimination rate constant (Kel= 0.0096±0.0013 hr⁻¹) and a prolonged half-life (T½= 73.84±11.32 h) relative to the free drug (Kel= 0.0122±0.0012 hr⁻¹, T½= 57.29±5.61 h), indicating a sustained systemic retention and slower clearance. Interestingly, while the Cmax of the raw miltefosine group (69.49±1.18 µg/ml) was slightly higher than that of the MF-ZnO-NPs group (59.76±7.36 µg/ml), this was offset by the extended exposure duration and improved bioavailability profile of the nanoparticle formulation. Collectively, these findings support the premise that MF-ZnO-NPs nanoparticles not only enhance the oral bioavailability of miltefosine but also modulate its pharmacokinetic profile in a manner that may contribute to prolonged therapeutic activity and improved safety.
Table 7: Summary pharmacokinetic parameters
| Summary of pharmacokinetic parameters | |||
| Parameters | Raw MF group A | MF-ZnO-NPs group B | |
| C max (µg/ml) | 69.48±1.18 | 59.755±7.36 | |
| T max (h) | 24 | 24 | |
| AUC0-t (h*µg/ml) | 4229.4±125.14 | 4820.5±160.5 | |
| AUC0-∞ (h*µg/ml) | 5539.7±240.99 | 7375.9±609.56 | |
| Kel (h-1) | 0.0122±0.0012 | 0.0096±0.0013 | |
| T ½ (h) | 57.293±5.61 | 73.837±11.32 | |
| % RB | 100% | 133.14% | |
*n=6 (Each Group)
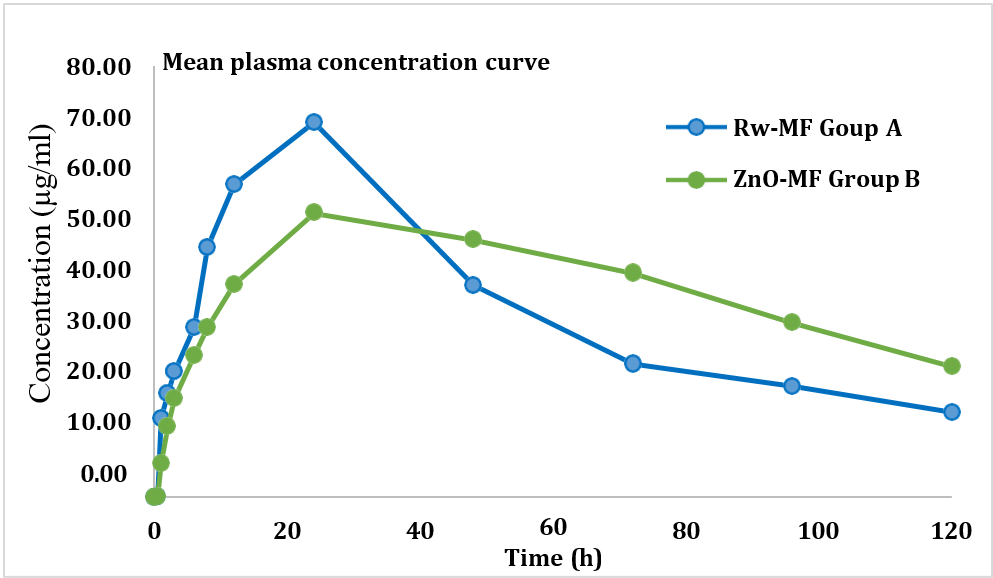
Fig. 6: Mean plasma concentration vs time curve
In vivo skin deposition study of miltefosine-loaded ZnO nanoparticles (24 h analysis)
The LC-MS/MS method was validated in skin homogenates for recovery and matrix effect. Extraction recovery was assessed by comparing analyte peak areas from pre-and post-extraction spiked samples and was found to be between 91.3% and 94.7%, confirming efficient and reproducible drug extraction. Matrix effects were evaluated using matrix factor calculations, which ranged from 0.93 to 1.07, with a %CV less than 10%, indicating minimal ion suppression or enhancement due to biological matrices. These parameters confirmed the method's reliability for quantifying miltefosine in skin samples. At the 24 h time point, the skin deposition analysis revealed notable differences between the two treatment groups. Group A, which received miltefosine-loaded ZnO nanoparticles (MF-ZnO-NPs), exhibited significantly higher drug accumulation in the skin tissue compared to Group B, treated with raw miltefosine. Specifically, the mean deposition observed in Group A was substantially elevated at 9.6±0.8 µg/g (n=3), reflecting a pronounced increase in localized drug retention. In contrast, Group B showed considerably lower skin deposition, measured at only 2.1±0.4 µg/g (n=6).
Table 8: Skin deposition (24 h analysis)
| Rat | Group A (MF-ZnO-NPs) µg/g | Group B (Raw miltefosine) µg/g |
| Rat 1 | 9.8±0.7 | 2.2±0.4 |
| Rat 2 | 9.5±0.8 | 2.0±0.3 |
| Rat 3 | 9.3±0.9 | 2.0±0.4 |
| Rat 4 | 9.7±0.8 | 2.1±0.3 |
| Rat 5 | 9.6±0.8 | 2.1±0.4 |
| Rat 6 | 9.6±0.9 | 2.2±0.4 |
| Mean±SD | 9.6±0.8 | 2.1±0.4 |
*n=6 (Each group)
These compelling results clearly underscore the capacity of MF-ZnO-NPs to markedly enhance the bioavailability of orally administered miltefosine, facilitating improved and sustained retention of the drug within the skin, thus potentially translating into enhanced therapeutic efficacy and reduced systemic exposure.
DISCUSSION
This study presents a comprehensive and multifaceted evaluation of miltefosine-loaded mesoporous ZnO nanoparticles (MF-ZnO-NPs), demonstrating significant improvements in pharmacokinetic behavior and safety profiles compared to conventional miltefosine. The biochemical, histopathological, pharmacokinetic, and deposition findings consistently underscore the benefits of nanoparticle encapsulation as a strategic platform for enhancing drug delivery and minimizing toxicity. The development of nanoparticulate systems for miltefosine delivery has been extensively explored to improve oral bioavailability and minimize systemic toxicity. Conventional delivery platforms, such as liposomal miltefosine formulations reported by Sundar et al., 2014, have demonstrated enhanced drug solubility and reduced gastrointestinal side effects due to their biocompatibility and ability to encapsulate hydrophobic molecules [22]. However, liposomes often suffer from limited drug-loading capacity, instability under gastrointestinal conditions, and high production costs, limiting their large-scale clinical application reported by Mendens et al., 2020 [23].
In comparison, the mesoporous ZnO nanoparticles (MF-ZnO-NPs) developed in this study offer several significant advantages. MF-ZnO-NPs provide a higher drug loading capacity (~60%), robust stability in simulated gastric and intestinal fluids, and a controlled biphasic drug-release profile with an initial burst followed by sustained release, ensuring both rapid onset and prolonged therapeutic exposure. Zhang et al., 2021 releveled the mesoporous structure reduces the accessible surface area of ZnO, limiting reactive oxygen species (ROS) generation, which is often associated with ZnO nanoparticle-induced cytotoxicity [24]. Furthermore, maintaining a controlled particle size (~178 nm) and administering optimized therapeutic doses contributed to the excellent biocompatibility observed in vivo.
Biochemical markers indicated a clear dose-dependent trend in hepatic and renal stress. The control and low-dose MF-ZnO-NPs groups maintained near-normal physiological parameters, whereas moderate to high doses of MF-ZnO-NPs, and notably the raw miltefosine group, demonstrated significant elevations in ALT, AST, creatinine, total bilirubin, and BUN. These biochemical findings align with previous observations on miltefosine-induced organ toxicity in other nanoformulations, such as liposomal carriers, which similarly show dose-dependent organ impairment reported by Carvalho et al., 2019 and Pérez-Victoria et al., 2006 [25, 26].
Histological analyses corroborated these biochemical outcomes, showing mild to moderate inflammatory and degenerative changes in hepatic and renal tissues of high-dose MF-ZnO-NPs groups, but far more severe alterations in the raw miltefosine group [27, 28]. The observed centrilobular necrosis, peri-glomerular inflammation, and epithelial disruption suggest a significant reduction in systemic toxicity with MF-ZnO-NPs at therapeutic dose levels.
In a behavioral assessment by Zhang et al., 2018, further supported the superior tolerability of MF-ZnO-NPs, as evidenced by maintained food intake and stable body weights in the low-and medium-dose groups. Conversely, raw miltefosine led to significant reductions in appetite and growth, consistent with biochemical and histopathological indicators of systemic toxicity [29].
Pharmacokinetic analysis demonstrated significant improvements in oral bioavailability with MF-ZnO-NPs, with an AUC0-inf increase from 5539.7±240.99 h·µg/ml (raw miltefosine) to 7375.9±609.56 h·µg/ml (MF-ZnO-NPs), reflecting a relative bioavailability of 133.14%. The prolonged half-life (73.84±11.32 h), reduced elimination rate, and sustained plasma retention indicate a favorable pharmacokinetic profile, consistent with advantages reported for liposomal miltefosine and other lipid-based nanocarriers [30, 31]. Notably, comparable Cmax and identical Tmax values suggest the nanoparticle formulation does not compromise absorption rate but significantly enhances drug exposure duration and extent. ZnO nanoparticles have been shown to inhibit efflux transporters like P-glycoprotein (P-gp), which limits drug extrusion from enterocytes back into the intestinal lumen [10]. Miltefosine, a known P-gp substrate, benefits from reduced efflux and increased transcellular absorption when encapsulated in ZnO nanoparticles.
Enhanced skin deposition results, demonstrating a nearly fourfold increase in localized drug retention with MF-ZnO-NPs compared to raw miltefosine at 24 h post-dose, are particularly clinically relevant in treating Post-Kala-Azar Dermal Leishmaniasis (PKDL). Increased cutaneous deposition potentially reduces systemic toxicity and improves therapeutic efficacy, contributing to enhanced patient compliance and reduced dosing frequency [32, 33].
Importantly, all pharmacokinetic and toxicity studies were consistently performed in rats, eliminating concerns regarding interspecies variability. However, the lack of long-term toxicity data beyond 28 days remains a limitation, necessitating future chronic toxicity studies to fully evaluate the long-term safety profile of the formulation.
CONCLUSION
The findings from this investigation Concludes that mesoporous ZnO nanoparticle-based delivery of miltefosine offers substantial therapeutic advantages over the conventional oral formulation. MF-ZnO-NPs nanoparticles significantly enhance oral bioavailability, prolong systemic circulation, and achieve greater skin deposition, all while attenuating the hepatic, renal, and gastrointestinal toxicities commonly associated with free miltefosine. The dose-dependent safety and efficacy observed in this study advocate for the potential clinical translation of MF-ZnO-NPs nanoparticles as a safer, more effective oral drug delivery system for miltefosine. These outcomes establish a promising foundation for further preclinical and clinical exploration, ultimately aiming to reposition miltefosine within a more biocompatible and pharmacologically optimized therapeutic framework.
ACKNOWLEDGEMENT
The authors would like to acknowledge ABS Clinical Solutions, Kolkata-152 and Jadavpur University, Kolkata-32 for the technical support and all other kinds of help provided for this study.
FUNDING
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sector.
AUTHORS CONTRIBUTIONS
P. G. (Parag Ghosh) contributed to conceptualization, nanoparticle synthesis, and methodology development. S. C. D. (Subas Chandra Dinda) supervised the study, guided the pharmacological evaluation, and critically reviewed the manuscript. D. D. (Debjit Dewan) assisted in LC-MS/MS method development and pharmacokinetic analysis. S. R. (Sukanta Roy) conducted the stability studies, in vitro drug release experiments, and histopathological analysis. D. Da. (Dibya Das) performed animal handling, dosing, and sample collection for pharmacokinetic and toxicity studies. S. D. (Sourav Das) carried out biochemical assays and assisted in data analysis and interpretation. A. B. (Anirbandeep Bose) provided project supervision, funding acquisition, and final approval of the version to be published. All authors contributed equally to the drafting, editing, and approval of the final manuscript.
CONFLICT OF INTERESTS
The authors hereby declare that they have no conflict of interest either to disclose.
REFERENCES
World Health Organization. Leishmaniasis. In: Geneva: World Health Organization; 2025. Available from: https://www.who.int/news-room/factsheets/detail/leishmaniasis. [Last accessed on 24 Apr 2025].
Palic S, Beijnen JH, Dorlo TP. An update on the clinical pharmacology of miltefosine in the treatment of leishmaniasis. Int J Antimicrob Agents. 2022;59(1):106459. doi: 10.1016/j.ijantimicag.2021.106459, PMID 34695563.
Dorlo TP, Balasegaram M, Beijnen JH, De Vries PJ. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J Antimicrob Chemother. 2012;67(11):2576-97. doi: 10.1093/jac/dks275, PMID 22833634.
Gu L, Lin J, Wang Q, Meng F, Niu G, Lin H. Mesoporous zinc oxide-based drug delivery system offers an antifungal and immunoregulatory strategy for treating keratitis. J Control Release. 2024 Apr;368:483-97. doi: 10.1016/j.jconrel.2024.03.006, PMID 38458571.
Anjum S, Hashim M, Malik SA, Khan M, Lorenzo JM, Abbasi BH. Recent advances in zinc oxide nanoparticles (ZnO NPs) for cancer diagnosis, target drug delivery and treatment. Cancers (Basel). 2021;13(18):4570. doi: 10.3390/cancers13184570, PMID 34572797.
Baskar G, Chandhuru J, Sheraz Fahad KS, Praveen AS, Chamundeeswari M, Muthukumar T. Anticancer activity of fungal L-asparaginase conjugated with zinc oxide nanoparticles. J Mater Sci Mater Med. 2015;26(1):5380. doi: 10.1007/s10856-015-5380-z, PMID 25589205.
Sathishkumar P, Li Z, Govindan R, Jayakumar R, Wang C, Long Gu FL. Zinc oxide quercetin nanocomposite as a smart nano-drug delivery system: molecular level interaction studies. Appl Surf Sci. 2021 Jan 15;536:147741. doi: 10.1016/j.apsusc.2020.147741.
Namvar F, Azizi S, Rahman HS, Mohamad R, Rasedee A, Soltani M. Green synthesis, characterization and anticancer activity of hyaluronan/zinc oxide nanocomposite. Onco Targets Ther. 2016 Jul 26;9:4549-59. doi: 10.2147/OTT.S95962, PMID 27555781.
Selvakumari D, Deepa R, Mahalakshmi V, Subhashini P, Lakshminarayan N. Anticancer activity of ZnO nanoparticles on MCF7 (breast cancer cell) and A549 (lung cancer cell). ARPN J Eng Appl Sci. 2015;10(12):5418-21.
Ahmad A, Imran M, Ahsan H. Biomarkers as biomedical bioindicators: approaches and techniques for the detection, analysis and validation of novel biomarkers of diseases. Pharmaceutics. 2023;15(6):1630. doi: 10.3390/pharmaceutics15061630, PMID 37376078.
Aryutova K, Stoyanov DS, Kandilarova S, Todeva Radneva A, Kostianev SS. Clinical use of neurophysiological biomarkers and self-assessment scales to predict and monitor treatment response for psychotic and affective disorders. Curr Pharm Des. 2021;27(39):4039-48. doi: 10.2174/1381612827666210406151447, PMID 33823771.
Chen XH, Huang S, Kerr D. Biomarkers in clinical medicine. IARC Sci Publ. 2011;163:303-22. PMID 22997869.
Ilyin SE, Belkowski SM, Plata Salaman CR. Biomarker discovery and validation: technologies and integrative approaches. Trends Biotechnol. 2004;22(8):411-6. doi: 10.1016/j.tibtech.2004.06.005, PMID 15283986.
North A, Ciaravino V, Mufti N, Corash L. Preclinical pharmacokinetic and toxicology assessment of red blood cells prepared with S-303 pathogen inactivation treatment. Transfusion. 2011;51(10):2208-18. doi: 10.1111/j.1537-2995.2011.03132.x, PMID 21985050.
Patel S, Patel S, Kotadiya A, Patel S, Shrimali B, Joshi N. Age-related changes in hematological and biochemical profiles of Wistar rats. Lab Anim Res. 2024;40(1):7. doi: 10.1186/s42826-024-00194-7, PMID 38409070.
Patel AG, Nariya MB, De S. Acute toxicity and repeated dose 28 d oral toxicity study of metriviv syrup in female rats. Ayu. 2018;39(2):107-12. doi: 10.4103/ayu.AYU_18_18, PMID 30783366.
Niyomchan A, Chatgat W, Chatawatee B, Keereekoch T, Issuriya A, Jaisamut P. Safety evaluation of the polyherbal formulation NawaTab: acute and subacute oral toxicity studies in rats. Evid Based Complement Alternat Med. 2023 Jul 24;2023:9413458. doi: 10.1155/2023/9413458, PMID 37528898.
Valicherla GR, Tripathi P, Singh SK, Syed AA, Riyazuddin M, Husain A. Pharmacokinetics and bioavailability assessment of miltefosine in rats using high-performance liquid chromatography tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2016 Sep 15;1031:123-30. doi: 10.1016/j.jchromb.2016.07.042, PMID 27475453.
Knudsen GA, Cheng Y, Kuester RK, Hooth MJ, Sipes IG. Effects of dose and route on the disposition and kinetics of 1-butyl-1-methylpyrrolidinium chloride in male F-344 rats. Drug Metab Dispos. 2009;37(11):2171-7. doi: 10.1124/dmd.109.029082, PMID 19704025.
Palic S, Chu WY, Sundar S, Mondal D, Das P, Pandey K. Skin pharmacokinetics of miltefosine in the treatment of post-kala-azar dermal leishmaniasis in South Asia. J Antimicrob Chemother. 2024;79(7):1547-54. doi: 10.1093/jac/dkae129, PMID 38727613.
Halder D, Das S, Ghosh B, Biswas E, Roy S, Bose A. An LC-MS/MS-based bioanalytical approach to resolve pharmacokinetic investigation of acotiamide hydrochloride and its application to bioequivalence study. Int J Pharm Pharm Sci. 2020;12(10):76-84. doi: 10.22159/ijpps.2020v12i10.38410.
Sundar S, Rai M. Advances in the treatment of leishmaniasis. Curr Opin Infect Dis. 2002;15(6):593-8. doi: 10.1097/00001432-200212000-00007.
Mendens A, Albuquerque P, Reis CP. Liposomes as a strategy to improve oral drug delivery: current issues and future perspectives. Int J Nanomedicine. 2020 Jun 20;15:9387-402.
Zhang J, Zhang F, Wu W, Gao Y. Controlled synthesis and applications of mesoporous ZnO nanoparticles. Adv Mater Interfaces. 2021;8(6):2001508.
Carvalho S, Cordeiro Da Silva A, Moreira J. Miltefosine: antitumor activity and clinical perspectives. Cancer Chemother Pharmacol. 2019;84(2):417-32.
Perez Victoria FJ, Castanys S, Gamarro F. Leishmania donovani resistance to miltefosine involves a defective miltefosine transporter. J Infect Dis. 2006;194(10):1382-8.
Luo Q, Jiang M, Kou L, Zhang L, Li G, Yao Q. Ascorbate conjugated nanoparticles for promoted oral delivery of therapeutic drugs via sodium-dependent vitamin C transporter 1 (SVCT1). Artif Cells Nanomed Biotechnol. 2018;46(Suppl1):198-208. doi: 10.1080/21691401.2017.1417864, PMID 29260899.
Khatik R, Dwivedi P, Shrinet V, Sahu D, Soni V. Miltefosine-loaded lipid nanocarriers for the effective management of leishmaniasis: formulation characterization and in vivo evaluation. Drug Dev Ind Pharm. 2014;40(9):1195-202.
Zhang W, Tanaka Y, Yamazaki M, Kawai Y, Kawai Y. Nanoparticle-based topical drug delivery systems for skin diseases. Nanomedicine (Lond). 2018;13(14):1639-51.
Rodrigues J, Dourado LF, Amaral MH, Silva AC. Lipid-based carriers for oral delivery of miltefosine: a review. J Drug Deliv Sci Technol. 2020;57:101689.
Pandey A, Singh S. Nanocarrier-based drug delivery systems for leishmaniasis treatment: an update. Curr Pharm Des. 2021;27(5):678-91.
Sharma R, Wadhwa S, Singh S, Lohan S. Topical nanocarrier systems for treatment of skin disorders: a review. Adv Pharm Bull. 2017;7(3):285-98.
Das S, Ali N. Nanotechnology-based treatment strategies against leishmaniasis. J Nanomed Nanotechnol. 2020;11(5):573.