
Int J App Pharm, Vol 17, Issue 6, 2025, 255-266Original Article
IMPROVED BIOAVAILABILITY OF ATORVASTATIN AND FENOFIBRATE THROUGH TERNARY SOLID DISPERSIONS: A POLYMER-SUPPORTED METHOD FOR THE ULTIMATE SOLUBILITY ENHANCEMENT
RAMESH BALASAHEB NAWALE1*, SANJEEV KUMAR SAHU1, MANISH VYAS1,2
1Lovely Professional University, Phagwara, Punjab-144411, India. 2Research and Development Cell, Parul University,Vadodara-391760,Gujarat, India.
*Corresponding author: Ramesh Balasaheb Nawale; *Email: nawale_ramesh@yahoo.co.in
Received: 02 May 2025, Revised and Accepted: 09 Oct 2025
ABSTRACT
Objective: This study aimed to improve the solubility and dissolution rate of a fixed-dose combination of atorvastatin (ATR) and fenofibrate (FEN) characterized by poor solubility via solid dispersion techniques.
Methods: Soluplus and Kollidon were used for formulating binary solid dispersion (BSD), and PVP and Kollidon were used for ternary solid dispersion (TSD) employing the solvent evaporation technique. Saturation solubility studies and in vitro drug release were done to quantify improvements in solubility and drug dissolution. Further characterization of solid dispersions was carried out by differential scanning calorimetry (DSC), scanning electron microscopy (SEM), X-ray diffraction (XRD), and Fourier-transform infrared spectroscopy (FTIR).
Results: Among binary systems, the SF8 formulation (drug-to-Soluplus® ratio of 1:4) showed an edge in solubilization enhancement, dissolution rate, and other physicochemical properties. The ternary composition KDF11 exhibited the best performance in terms of solubility and dissolution rate. In vivo, pharmacokinetic studies reaffirmed that drug bioavailability was greatly improved. SF8 BSD increased solubility by 17-fold. ATR solid dispersion produced 2.87 times more Cmax and 2.56 times more AUC₀–₂₄ₕ while FEN nanoformulation produced 3.56 times more Cmax and 3.44 times more AUC₀–₂₄ₕ after their pure drug counterparts. Cmax increased 2.87-fold for ATR. These increases can be understood to be attributed to the conversion of the crystalline structure into an amorphous state, thereby enhancing solubility and facilitating a more favorable concentration gradient across gastrointestinal and systemic barriers.
Conclusion: The study demonstrated that in the ternary system, solid dispersion formulations were very much better than binary dispersions in improving the solubility and dissolution of poorly soluble drug combinations. TSD offers a viable strategy for enhancing FDC bioavailability with potential application in the effective management of hyperlipidemia.
Keywords: Atorvastatin, Polyvinyl pyrrolidone, Fenofibrate, Binary solid dispersion, Ternary solid dispersion, Soluplus, Kolliphor® P 407, Kollidon, Pharmacokinetics
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i6.54838 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
When two or more active pharmaceutical ingredients (APIs) in a single dosage form make up fixed-dose combinations or FDCs. Particularly in chronic diseases like cardiovascular disease, lipid disorders, and metabolic syndrome, oral FDCs have attracted interest in simplifying medication regimens and improving patient adherence, given an aging population and growing lifestyle-related diseases. Furthermore, improving solubility, dissolution, and bioavailability through formulation changes could help to lower the possible dosage. FDCs thus maximize medication efficiency and help to lower healthcare resource use [1].
Hyperlipidemia is a common condition found in older people who have type 2 diabetes and metabolic syndrome. Hypertriglyceridemia, elevated low-density lipoprotein cholesterol (LDL-C) levels, and decreased high-density lipoprotein cholesterol (HDL-C) levels are all common lipid disorders facing this group. If statins prove ineffective, combination therapy using fibrates is considered. Fenofibrate (FEN) is a prodrug that metabolizes in the liver into fibric acid, stimulating peroxisome proliferator-activated receptor-alpha (PPAR-α) [2]. Fibrates act on this pathway to enhance lipolysis, stimulate lipoprotein lipase, and encourage apolipoprotein A-I and A-II synthesis, resulting in lowering triglycerides (TG) and LDL-C levels while raising HDL-C. The major therapeutic target is the lowering of LDL-C, along with lowering high TG and low HDL-C [3].
As fibrates are capable of significantly lowering TG and increasing HDL-C, management of lipid disorders becomes paramount in the antihypertensive regimen. FEN is favored as an add-on agent when statins alone are inadequately efficacious, as it carries a lesser risk of inducing myopathy. Atherogenic patients may benefit from FEN-statin combinations since DIACOR and ACCORD are among the studies that show enhanced lipid improvements associated with FEN-statin combinations over single-drug therapy. Clinical studies have shown that combination therapies produce greater reductions in total cholesterol, TG, and low-density lipoprotein cholesterol levels compared with statin monotherapy [4].
Target LDL-C achievement rates with the combination have improved, particularly in high-risk patients or those with intolerance to high-dose statins. As per the ESC guidelines, FEN does not affect the metabolism of statins and does not increase the risk of statin-induced myopathy.
Research shows that FEN and Simvastatin (SIM) combine to create an eutectic system that improves dissolving characteristics. Oral FDCs, which are frequently used to treat conditions, may benefit from this advancement. Despite the existence of SIM and FEN FDCs, research on their FDCs as solid dispersion is a little less explored.
Agata Górniak et al. in 2023 reported eutectic solid dispersions of drug FEN and SIM by kneading with ethanol. Still, no studies are showing the binary or ternary solid dispersions (TSD) using these drugs [5]. These techniques have been adapted with the hope of improving solubility for these Biopharmaceutical Classification System (BCS) Class II drugs that are practically insoluble: FEN (0.1 µg/ml) and SIM (30 µg/ml) possess significantly higher solubility compared with SIM. Solid dispersion of FDC of FEN and ATR was reported by Sobhita Rani et al. using Polyethylene glycol 4000 and 6000, and the authors have reported an increase in solubility and dissolution, but they did not prove the oral bioavailability of the same [6].
Hence, to enhance the dissolution and absorption, many efforts were made by many scientists using solid dispersion, separately and in combination, binary solid dispersions (BSD) and TSD for the selected drugs were not reported [7].
To our knowledge, this is the first report of TSD for an ATR-FEN fixed-dose combination using a synergistic blend of Soluplus, Kollidon VA64, and PVP K30. This unique combination stabilises the amorphous drug form, inhibits recrystallisation, and improves wettability and solubilisation via these polymers' complementary processes. Soluplus, an amphiphilic polyvinyl caprolactam–polyvinyl acetate–polyethylene glycol graft copolymer, improves medication wettability and forms stable amorphous systems. The copovidone Kollidon VA64 is widely utilised for its film-forming and drug recrystallization-preventing properties. Hydrophilic polymer PVP K30 is used to improve dissolving by hydrogen bonding and solubilisation. These polymers were chosen because of their efficacy in earlier solid dispersion formulations and their synergistic ability to address ATR and FEN solubility issues in a combined system.
Amorphous solid dispersions (ASDs) are those solid-state systems in which one or several hydrophobic APIs are dispersed molecularly within a wide range of hydrophilic polymer matrices [8]. These are well-known formulations that improve the solubility and dissolution rates of poorly water-soluble drug substances. Generally, this amorphous transformation increases thermodynamic solubility and enhances bioavailability [9-13].
Hence, this study was taken up to evaluate the potential of BSD and TSD of the selected fixed drug combination to enhance the solubility and dissolution, aiding in the development of an oral fixed-dose formulation with improved bioavailability and stability.
MATERIALS AND METHODS
Materials
ATR and FEN were given as gift samples from Innomedix Pharma Pvt Ltd., Jaipur, India. Soluplus®, Kollidon® VA 64, PVPK-30 (polyvinyl pyrrolidone), and Kolliphor® P 407® P 407 were provided by TCI Chemicals, India. All the solvents were of analytical grade.
Methods
Formulation of solid dispersion
The solid dispersion was prepared using the solvent evaporation method. Using different drug-to-polymer ratios, the various BSDs were prepared using Soluplus and Kolliphor® P 407® P 407. The TSD of a binary formulation has been refined with the inclusion of polymers, namely PVP and Kollidon. Solid dispersions in binary and ternary configurations can be found in table 1. The right amount of methanol (20 ml) was used to dissolve 120 mg of ATR and 840 mg of FEN, both of which were precisely measured. In a separate 20 ml of methanol, the chosen carrier was dissolved while being constantly stirred. For a more consistent mixture, these two solutions were sonicated for 10 min. An apparatus for rotational evaporation was used to evaporate the solvent at a temperature of 45 °C, ~150 mbar pressure, and 120 rpm rotation speed. We dried the end result at 40 °C for 24 h. We dried the result after pulverization and then ran it through a No. 80 mesh screen. For future use, the powder was sealed in a bottle with desiccants [7].
A TSD was created by combining 30 ml of the obtained BSD with 20 ml of the selected carrier (Kollidon or PVP) dissolved in methanol. Sonication was used to combine the two solutions. The next procedures for preparation were identical to those for making a BSD. After combining the enhanced TSD with microcrystalline cellulose, sodium starch glycolate, and a tablet-forming punching machine, a dispersible tablet was produced.
Evaluation of BSD and TSD
Solubility studies
The solubility of unadulterated medicines (ATR and FEN), together with optimised BSD and TSD formulations, was assessed with the shake-flask method. Precisely measured quantities of each formulation, comprising 2 mg of atorvastatin and 10 mg of fenofibrate, were individually introduced into 10 ml of phosphate buffer at pH 7.4 and simulated stomach fluid at pH 1.2. The selection of pH 1.2 and pH 7.4 was intended to replicate gastric and intestinal environments, respectively, to evaluate drug solubility under physiological pH levels pertinent to oral absorption. Each sample was agitated and thereafter positioned in a thermostatically regulated shaker at 37 ± 0.5 °C and 100 rpm for 48 h to achieve equilibrium. Samples were collected at 24 and 48 h, and the consistency in drug concentration between these two time points was utilised to validate equilibrium solubility. The solutions were centrifuged at 10,000 rpm for 15 min post-shaking to isolate the undissolved medication. The supernatants were filtered using a 0.45 μm membrane filter, suitably diluted, and analysed with a UV-visible spectrophotometer at λmax 246 nm for ATR and 286 nm for FEN.
Furthermore, solubility and dissolution studies were performed in a 0.5% sodium lauryl sulphate (SLS) aqueous solution, a widely recognised surfactant system for poorly water-soluble BCS Class II medicines. The application of 0.5% SLS aids in sustaining sink conditions and enhancing discriminatory power in dissolution testing. In response to the reviewer's recommendation, we recognise the significance of physiologically relevant conditions and have incorporated comparative solubility testing in biorelevant media, specifically FaSSIF (fasted state simulated intestinal fluid) and FeSSIF (fed state simulated intestinal fluid), to enhance the prediction of in vivo performance [14].
Practical yield and drug content
The yield of the formulation was determined by taking the difference between dried BSDS and TSD (W1) and the original mass of the drugs and polymers taken(W2). The percentage practical yield is calculated using the following formula:

For the determination of drug contents, an equivalent amount of BSD and TSD formulations containing 2 mg of ATR and 10 mg of FEN was mixed with 10 ml of organic solvent, like methanol, and subjected to 10 min of sonication to ensure the complete solubility of the drug. After suitable dilutions, the samples were filtered and analyzed by using a UV-visible spectrophotometer at a lambda max of 246 nm (ATR) and 286 nm (FEN) [15].
Drug release
For determining drug release from BSDF, TSDF, TSDF-T, and plain drug, USP dissolution (Type 2) was employed. Pure drugs and SDs containing an equivalent amount of drugs (20 mg of ATR and 120 mg of FEN) were weighed accurately and added into the dissolution vessels containing 0.5 % Sodium lauryl sulfate of 900 ml (pH 7.4). The dissolution was carried out at 37±0.5 °C at an rpm of 50. At regular time intervals, samples were taken, filtered and analyzed, and calculated and plotted the percentage of drug release [7].
SEM (Scanning electron microscopy)
The surface morphology of pure drugs (ATR, FEN), BSD, and TSD formulations was examined using Field Emission Scanning Electron Microscopy (FESEM) (Carl Zeiss, Germany). Before analysis, the samples were affixed to aluminium stubs with double-sided carbon adhesive tape and coated with a thin layer of platinum (approximately 10 nm) using a sputter coater (Quorum Technologies) set at 20 mA for 40 seconds to enhance surface conductivity. Imaging was conducted in a vacuum at an accelerating voltage of 10 kV, with micrographs captured at different magnifications to evaluate particle shape, aggregation, and surface texture.
Compatibility studies
To assess potential interactions between pharmaceuticals and polymers, Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) spectroscopy was performed utilising a Bruker Alpha II spectrometer. Approximately 5 mg of each material (pure medicines, polymers, BSD, and TSD) was immediately deposited onto the ATR crystal without additional processing. Spectra were obtained throughout the range of 4000–400 cm⁻¹ at a resolution of 4 cm⁻¹, averaging 16 scans for each sample. Key functional group alterations were examined to evaluate drug-polymer compatibility.
DSC study (Differential Scanning Calorimeter)
Thermal analysis of ATR, FEN, polymers, BSD, and TSD formulations was conducted utilising a PerkinElmer DSC 4000 instrument. Samples weighing 2–5 mg were enclosed in aluminium pans and subjected to heating from 30 °C to 200 °C at a rate of 5 °C/min, maintained under a continuous nitrogen purge with a flow rate of 20 ml/min. A reference was established using an empty aluminium pan. The lack or expansion of specific drug melting endotherms was interpreted as an indication of amorphous conversion and effective dispersion within the polymer matrix.
XRD (X-ray diffraction)
The crystallinity of pure drugs, physical mixtures (PM), BSD, and TSD was assessed using a Philips PW-1710 X-ray diffractometer, which utilised a Cu Kα radiation source (λ = 1.5406 Å) and a graphite monochromator. Scans were conducted within the 2θ range of 2° to 80°, utilising a step size of 0.045° and a counting duration of 0.5 sec per step. The instrument operated at 40 kV and 30 mA. Samples were finely ground to achieve a uniform particle size prior to placement on the sample holder. The absence or diminished intensity of characteristic drug peaks indicates a decrease in crystallinity or a transition to an amorphous state.
HPLC method
The concentrations of FEN and ATR were ascertained utilizing a Water HPLC System equipped with a UV-DAD detector with slight modification as reported by Jain et al. The mobile phase was acetonitrile and water in the ratio of 70:30 v/v. A C18 column RP-18, of 125 mm x 3 mm, 5 microns, was used for the study. The samples were eluted at a flow rate of 1.0 ml/min, with FEN spotted at wavelengths of 286 nm and 238 nm. The retention times of FEN and SIM were 3.4 and 2.3 min. The standard calibration curve in the range of 0.3 to 250 µg/ml and 0.2 to 200 µg/ml with correlation values over 0.999 was plotted graphically. For the detection of drugs in the plasma specimen, the protein precipitation method was utilized. The drugs were separated from serum (50 µl**) by mixing 300 µl** of acetonitrile (ACN), mixing the contents for 10 min, and then subjected to centrifugation for 12 min at 8000 rpm before analysis by high-performance liquid chromatography (HPLC) [16].
Exploration of pharmacokinetic parameters
The research involved healthy male Wistar rats aged 4–5 w, with a body weight of 200±20 g, obtained from the National Institute of Nutrition (NIN), Hyderabad, India. All experimental procedures received approval from the Institutional Animal Ethics Committee (IAEC) under Protocol No. 1447/PO/Re/S/11/CPCSEA-102/A, and the study complied with the guidelines set forth by the Committee for Control and Supervision of Experiments on Animals (CPCSEA). Rats were acclimatised for 7 days in a controlled environment, adhering to standard laboratory conditions. (Temperature: 20±2 °C; Humidity levels range from 40% to 60%; Light/Dark cycle: 12 h of light and 12 h of darkness; Diet consists of standard pellet chow and water provided ad libitum).
Animals were randomly assigned to two groups (n=6 per group). Group I (Control) received a suspension of pure ATR and FEN in 0.5% w/v sodium carboxymethylcellulose (CMC). Group II (Test): Administered the optimised TSD formulation comprising atorvastatin (8 mg/kg) and fenofibrate (20 mg/kg). The oral dose of ATR (8 mg/kg) was determined using human equivalent dose scaling, following FDA-recommended interspecies dose conversion factors, and corresponds with standard therapeutic dosing in clinical practice. The FEN dose of 20 mg/kg was selected to align with established preclinical pharmacokinetic studies for lipid-lowering agents. Blood Sampling and Analysis: Serial blood samples (200 µl** each) were obtained from the retro-orbital venous plexus at specified time intervals: 0.25, 0.5, 1, 2, 4, 6, 8, and 12 h following administration. The volume of blood collected was restricted to a maximum of 10% of the total circulating volume per week, in compliance with animal welfare guidelines to prevent stress or anaemia. Samples were collected in EDTA-coated tubes, centrifuged at 7500 rpm for 10 min at 4 °C, and the plasma was separated and stored at −20 °C until analysis. Plasma drug concentrations were measured utilising a validated HPLC method. Pharmacokinetic parameters such as Cmax, Tmax, AUC₀–₁₂, and t₁/₂ were calculated using a non-compartmental analysis model with Kinetica 5.0 software. Statistical comparisons were conducted utilising GraphPad Prism version 8.05, with results presented as mean±SD.
Stability studies
Batches of optimized BSD (SF8) and TSD (KDF11) were made using the solvent evaporation method as described in the formulation section. The samples were maintained for three months in two conditions: refrigerated at 4±2 °C (dark, no humidity control) and at ambient temperature (25±2 °C/60±5% RH) (ICH long-term-like setting). Sampling was performed at the initial time point (T₀) and after three months (T₃). All analyses were performed under low-light conditions, with each condition and time point examined in triplicate (n = 3) for each analyte and formulation, unless otherwise stated.
Statistical analysis
All experimental data were presented as Mean±Standard Deviation (SD). One-way Analysis of Variance (ANOVA) was utilised to conduct statistical comparisons among various formulation groups. A p-value below 0.05 (p<0.05) is deemed statistically significant. Statistical analyses were conducted utilising GraphPad Prism software (version 8.05, GraphPad Software Inc., CA, USA).
RESULTS AND DISCUSSION
Formulation of BSD and TSD
For the FDCs, the BSD using two polymers, such as Soluplus and Kolliphor® P 407® P 407 were used in drug-to-polymer proportions, and from the enhanced BSD, the TSD were formulated with two polymers, such as Kollidon and PVP, at ratios, and the compositions of the BSD and TSD are given in table 1.
Table 1: Composition of BSD and TSD (w/w)
| Formulation code (BSD) | Drug: polymer (w/w) | Drug: polymer (weight in mg) |
| KF1 | 1:1 | 960 mg: 960 mg |
| KF2 | 1:2 | 960 mg: 1920 mg |
| KF3 | 1:3 | 960 mg: 2880 mg |
| KF4 | 1:4 | 960 mg: 3840 mg |
| SF5 | 1:1 | 960 mg: 960 mg |
| SF6 | 1:2 | 960 mg: 1920 mg |
| SF7 | 1:3 | 960 mg: 2880 mg |
| SF8 | 1:4 | 960 mg: 3840 mg |
| Formulation code (TSD) | Drug: Kollidon®VA 64: Ternary polymer | Drug: kollidon®VA 64: ternary polymer |
| KDF9 | 1:4:1 | 960 mg: 3840 mg: 960 mg |
| KDF10 | 1:4:2 | 960 mg: 3840 mg: 1920 mg |
| KDF11 | 1:4:3 | 960 mg: 3840 mg: 2880 mg |
| PVPF12 | 1:4:1 | 960 mg: 3840 mg: 960 mg |
| PVPF13 | 1:4:2 | 960 mg: 3840 mg: 2880 mg |
| PVPF14 | 1:4:3 | 960 mg: 3840 mg: 2880 mg |
Note: 120 mg of ATR and 840 mg of FNF was taken in each BSD and TSD
Evaluation of BSD and TSD
Solubility studies
The solubility of the pure drugs was evaluated in distilled water, 0.1 N HCl (pH 1.2), and pH 7.4 phosphate buffer. The ATR solubility is 3.02±0.09 µg/ml, 19.78±3.66 µg/ml, and 4.72±0.57 µg/ml, whereas FEN solubility is 5.89±0.84 µg/ml, 42.09±5.31 µg/ml, and 54.86±4.43 µg/ml. ATR has a little higher solubility in 0.1 N HCl, whereas FEN has high solubility in phosphate buffer.
The solubility profiles of the formulated BSD and TSD are shown in fig. 1 and 2.
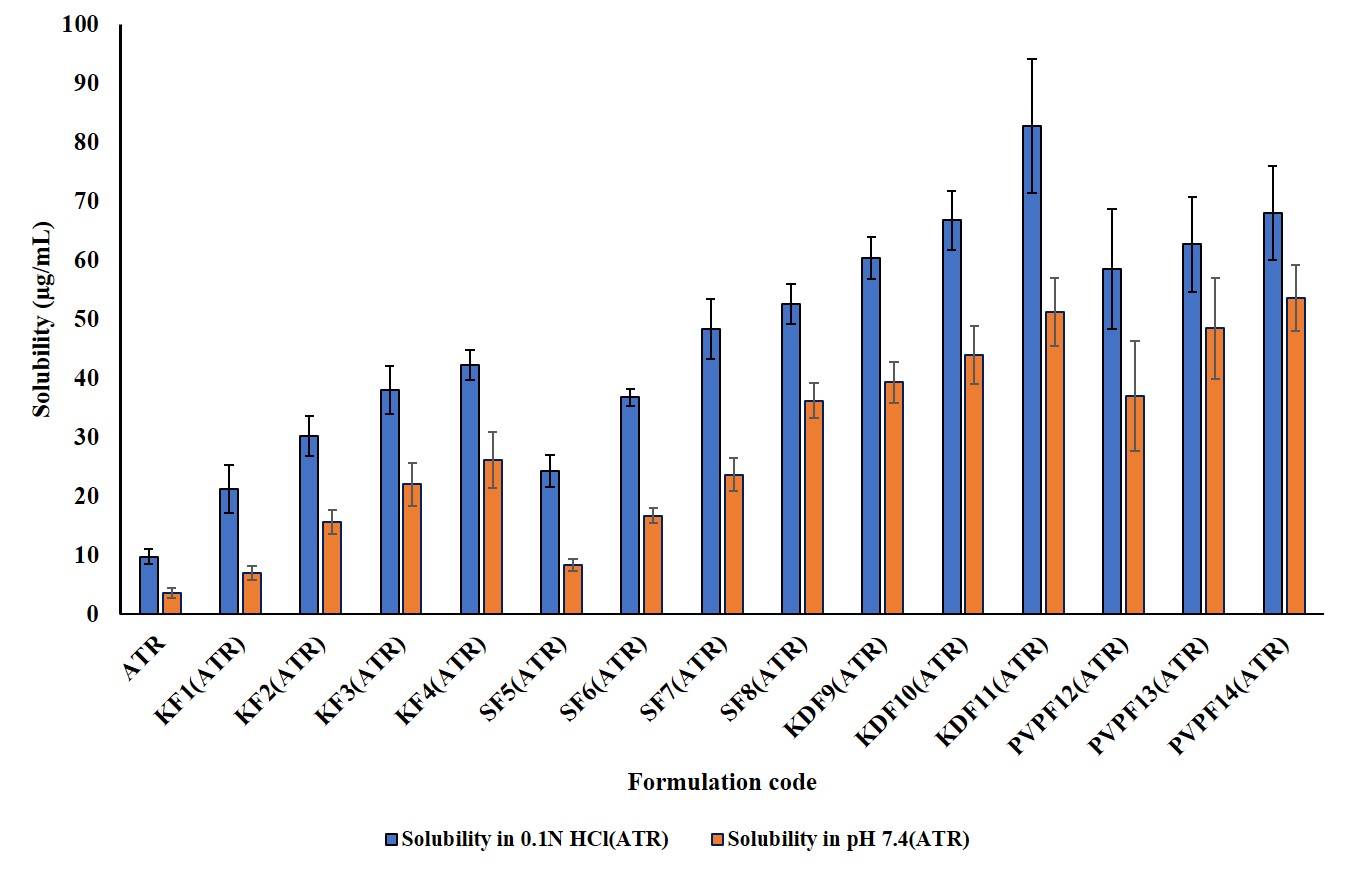
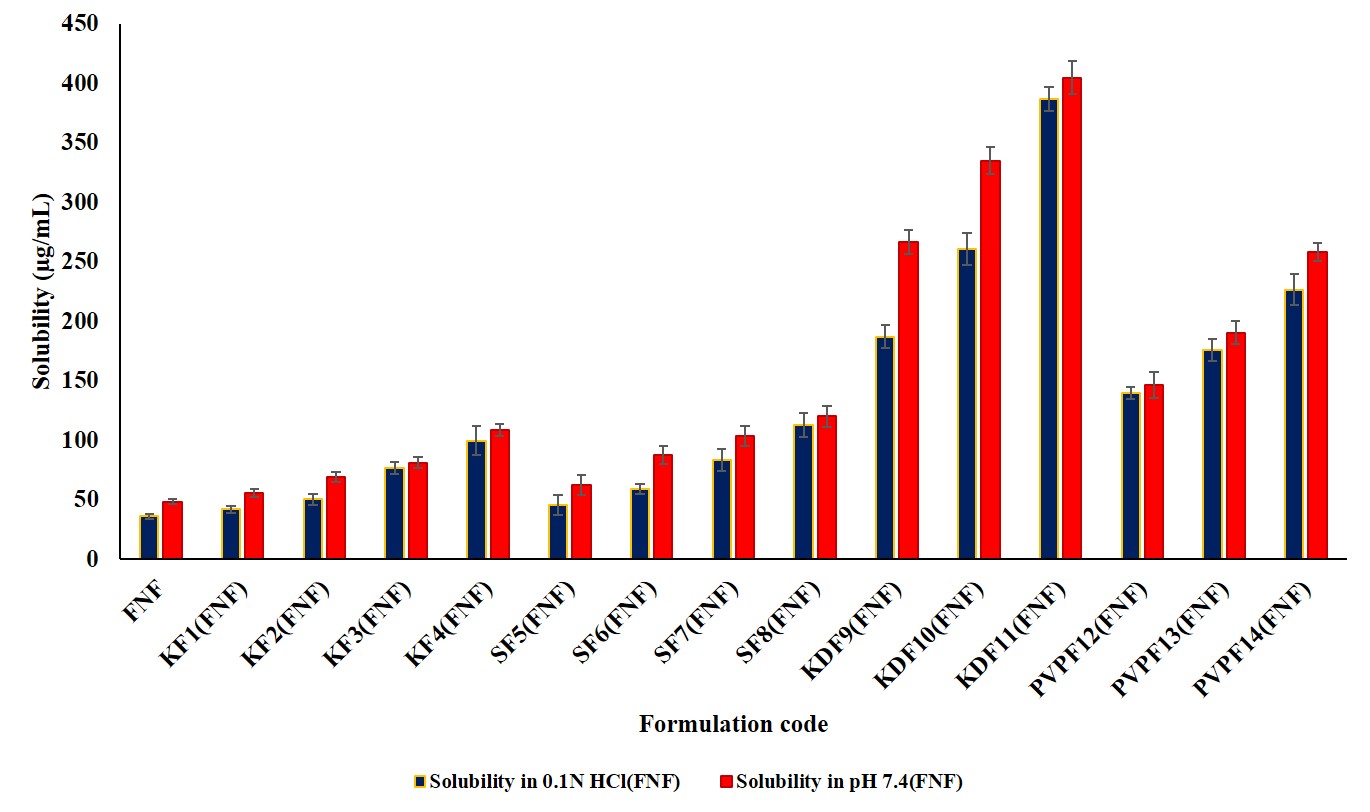
Fig. 1 and 2: Solubility studies of ATR and FEN in BSD and TSD along with plain drugs. Error bars indicate SD of triplicate
Among ATR BSDs, the SF8 formulation had the greatest saturation solubility, measuring 52.56±3.40 μg/ml in 0.1 N HCl and 36.24±2.9 μg/ml in pH 7.4 phosphate buffer (PB). Conversely, the KDF11 TSD demonstrated the most significant increase in solubility, attaining 82.84±11.4 μg/ml in 0.1 N HCl and 51.28±5.7 μg/ml in pH 7.4. The substantial enhancement in solubility is ascribed to the whole transformation of the drug from its crystalline to amorphous form. In the context of FEN BSD, the SF8 formulation exhibited the maximum solubility, measuring 112.72±10.24 in 0.1 N HCl and 120.22±8.96 μg/ml in pH 7.4. The KDF11 (TSD) TSD demonstrated the most significant increase in solubility, attaining 386.56±10.16 in 0.1 N HCl and 404.77±13.64 μg/ml in pH 7.4.
Soluplus is an amphiphilic polymer that can be used for the enhancement of wetting for better drug dispersibility. It could generate micellar structures and hydrogen bonding to keep the amorphous substance from recrystallizing. The polymer matrix makes drug-polymer miscibility better on the molecular level dispersion due to the dispersion of the polymeric matrix [17].
Thus, it would lead to improved solubility perception of the drug, although the drug is less soluble in water. To improve solubility, the system of binary drug-Soluplus is further expanded by adding Kollidon® to a TSD. Kollidon not only works as an additional ingredient but also functions as a hydrophilic carrier to increase the wettability and dispersibility of the drug formulation [8]. Further, it would stabilize the amorphous state, lowering the chances of recrystallization as it establishes hydrogen bonding with the drug and Soluplus. The combined contribution of Soluplus and Kollidon improves drug-polymer miscibility and supersaturation maintenance. Kollidon also improves the porosity and surface area of the dispersion, which speeds up the dissolution of the medicine. All these mechanisms act synergistically toward improving the solubility of these not-very-water-soluble ternary-system medicines [8].
Drug content and practical yield
The drug content of the various BSD and TSD was determined for ATR and FEN, and in the formulations, the drug content varied from 96.02±5.49 to 89.15±2.12 in the case of ATR, and it varied from 94.35±5.72 to 89.06±3.06. It can be said that a good amount of the drug has been dispersed in the selected carrier, and in all the formulations, the practical yield is more than 95 percent (not shown).
Table 2: Data on drug content of ATR and FEN in BSD and TSD
| Formulation code (BSD) | Drug content of ATR in BSD and TSD | Drug content of FEN in BSD and TSD |
| KF1 | 92.24±4.23 | 93.12±2.36 |
| KF2 | 93.44±2.45 | 94.27±3.83 |
| KF3 | 89.15± 2.12 | 92.8±4.04 |
| KF4 | 90.37±3.05 | 91.33±2.41 |
| SF5 | 95.88±5.86 | 94.26±5.29 |
| SF6 | 94.04±3.27 | 89.06±3.06 |
| SF7 | 94.56±4.86 | 91.67±4.64 |
| SF8 | 91.23±3.66 | 90.81±2.30 |
| KDF9 | 91.34±4.10 | 92.22±4.18 |
| KDF10 | 93.45±3.33 | 91.98±4.69 |
| KDF11 | 93.56±5.74 | 94.35±5.72 |
| PVPF12 | 90.89±2.63 | 91.58±4.36 |
| PVPF13 | 96.02±5.49 | 92.39±4.44 |
| PVPF14 | 94.66±5.89 | 91.59±2.49 |
Value expressed as mean±SD, n=3
Drug release
From the results, it is revealed that the pure drug released only 29.31±3.44 in PB pH 7.4 after 2 h in the case of ATR, whereas the FEN pure drug released only 20.26±2.14 in phosphate buffer. In the first half an hour, only 11.48±4.91 percent of ATR and 10.24±1.03 percent of FEN were released. However, the BSD released more than 86 percent of the drug and TSD released more than 99 percent of the drug released in 2 h from all the formulations, and the maximum drug released was from SF8 showing 86.25±5.56percent of drug release, and KDF11 released 98.44 ±2.56percent in case of ATR from phosphate buffer pH 7.4. While FEN has released 91.72±6.27 from BSD and 99.25 ±3.07inphosphate buffer. Overall, all the formulations have shown an increased drug release in comparison to the plain drugs. The reason is owing to the transformation of the crystalline form to the amorphous form, which increased the wettability of the drug. The drug release from ATR and FEN is shown in fig. 3 and 4.

Fig. 3: In vitro drug release from BSD (A, B), TSD (C, D), and plain drugs from different formulations. Error bars indicate SD of triplicate
The BSD formulated with Soluplus has increased drug wettability; the mechanism is the conservation of supersaturation resulting from intermolecular hydrogen bonding between the drug and the soluplus. As compared to the BSD prepared with Kolliphor® P 407, Soluplus has increased the drug release. Soluplus also has a swelling ability, which also helps in fast drug release from the dispersion [15, 17].

Fig. 4: In vitro drug release from BSD (E, F), TSD (G, H), and plain drugs from different formulations. Error bars indicate SD of triplicate
The solubility and drug release were further increased in TSD in the case of Kolliphor® P 407 and the reason can be attributed to the surfactant property of the polymer, it is mainly composed of ethylene oxide and propylene oxide blocks and has the nature of self-assembling ability into micelle when placed in aqueous media, which therefore enhances the solubilization of the drug [18]. Therefore, based on the results, TSD formulated with Kolliphor® P 407 was further selected for further characterization, stability, and in vivo studies.
SEM (Scanning electron microscopy)
Fig. 5 shows the formulation's and the simple drug's exterior surface characteristics. In its authentic state, the drug has a broad particle size range with distinct units and an irregular cubic form with micrometer-sized particles. In addition, the BSD with Soluplus and Kollidon showed the disappearance of crystalline as the drug was masked by the polymer coating, and the results are very well supported by XRD studies [19].

Fig. 5: SEM pictures of A) and B) pure drug of ATR and FEN, C) BSD with Kolliphor® P 407; D) BSD with Soluplus; E) TSD with Kollidon; F) TSD with PVP
Compatibility studies
Attenuated total reflection fourier transform infrared spectroscopy (ATFT-IR)
Fig. 6 and table 3 depict the determination of component compatibility through IR spectra analysis of the BSD and TSD, excipients, and drugs. The range of 400-4000 cm⁻¹ was used to record the FTIR spectra. In the FTIR spectra of FEN, a variety of absorption bands could be identified, such as the C-H stretch (2984 cm⁻¹), ester carbonyl stretches (1725 cm⁻¹), carbonyl stretch (1648 cm⁻¹), benzene ring stretch (1597 cm⁻¹), and aryl ether (1286 cm⁻¹). Likewise, the SIM showed absorption bands corresponding to stretching of O-H (3547 cm⁻¹), aliphatic C-H vibration (2951, 2930, 2872, and 1467 cm⁻¹), ester C=O vibrations (1695 and 1266 cm⁻¹), and ester/lactone C-O-C oscillations (1162 cm⁻¹). The FTIR spectra of solid dispersion reflected a superimposition of bands from both drugs, and no additional bands or significant shifts were observed, confirming the absence of chemical interactions among the formulation components [20, 21].
Table 3: FTIR peak assignments
| Wavenumber (cm⁻¹) | Assignment | Observed in |
| 3547 | O–H stretching | ATR |
| 2984 | C–H stretching | FEN |
| 2951, 2930, 2872 | Aliphatic C–H stretching | ATR |
| 1725 | Ester carbonyl (C=O) stretch | FEN |
| 1695 | Ester carbonyl stretch | ATR |
| 1648 | Ketone carbonyl (C=O) stretch | FEN |
| 1597 | Aromatic benzene ring stretch | FEN |
| 1467 | CH₂ bending | ATR |
| 1286 | Aryl ether (C–O) stretch | FEN |
| 1266 | Ester C–O stretch | ATR |
| 1162 | Ester/lactone C–O–C oscillations | ATR |
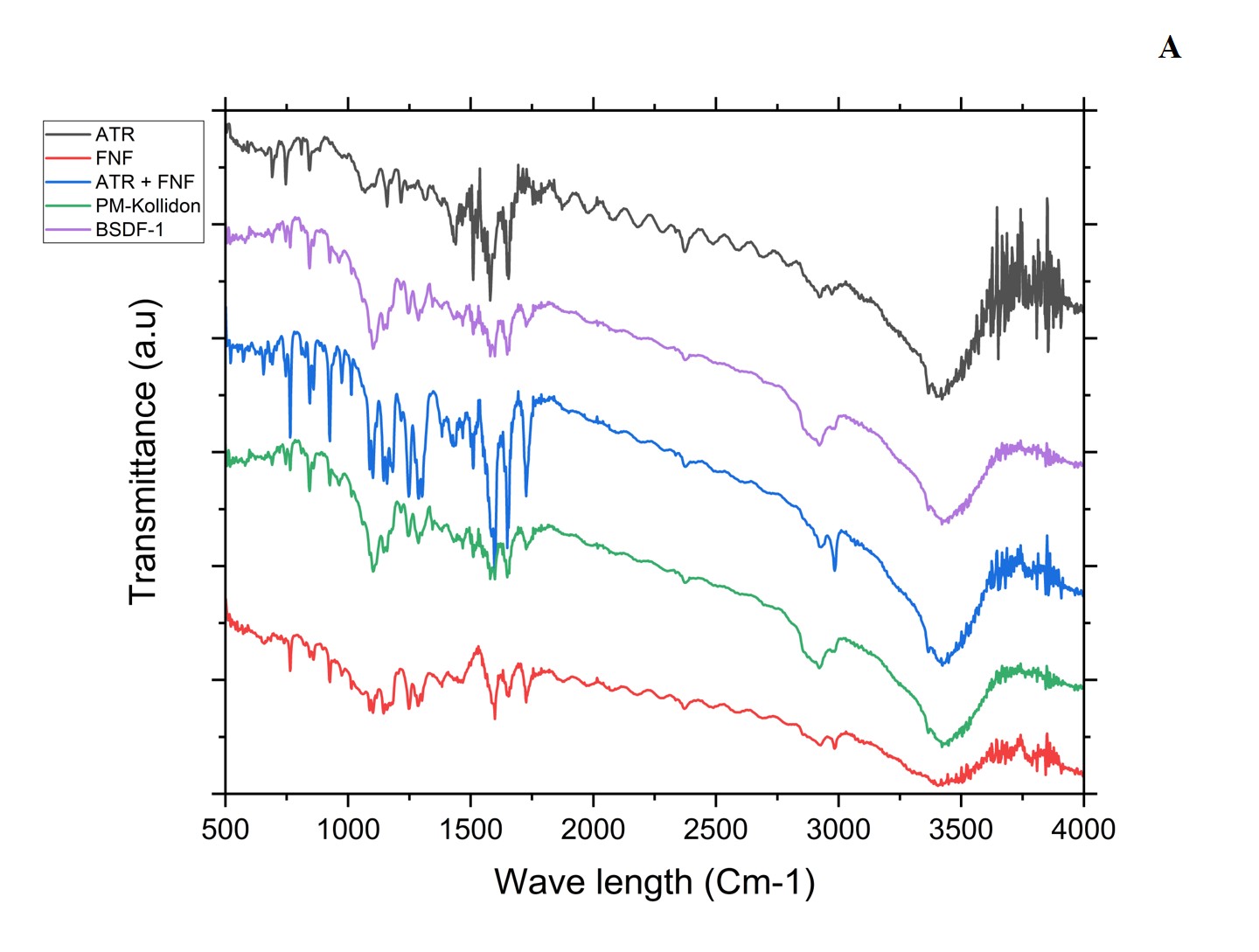

Fig. 6A and B: ATFT-IR of plain drugs (ATR, FEN), physical mixture, BSD, and TSD along with PM and combination plain drugs
DSC Study (Differential Scanning Calorimetry)
The thermal behavior of the optimized BSD, TSD, the physical mixture, and the pure drugs according to DSC is portrayed (fig. 7). The ATR PD was reported to have a melting point of 128.18 and 240.23 °C. The exceedingly crystalline structure of FEN PD was evidenced by its sharp melting endotherm at 83.65 °C. The drugs have shown a characteristic peak in the physical mixture. Nonetheless, in BSD, an endothermic peak at 84.03 °C of the FEN with a reduced crystallinity and another peak at 237.28 °C, indicating that ATR has undergone a significant amorphous nature and might have been covered by stabilizer acting as an inhibitor for further crystal growth. Since no new additional peaks are observed, we can confirm that the drugs are chemically stable and no degradation products were formed, and the same is reported by Agata Górniaketal [5].
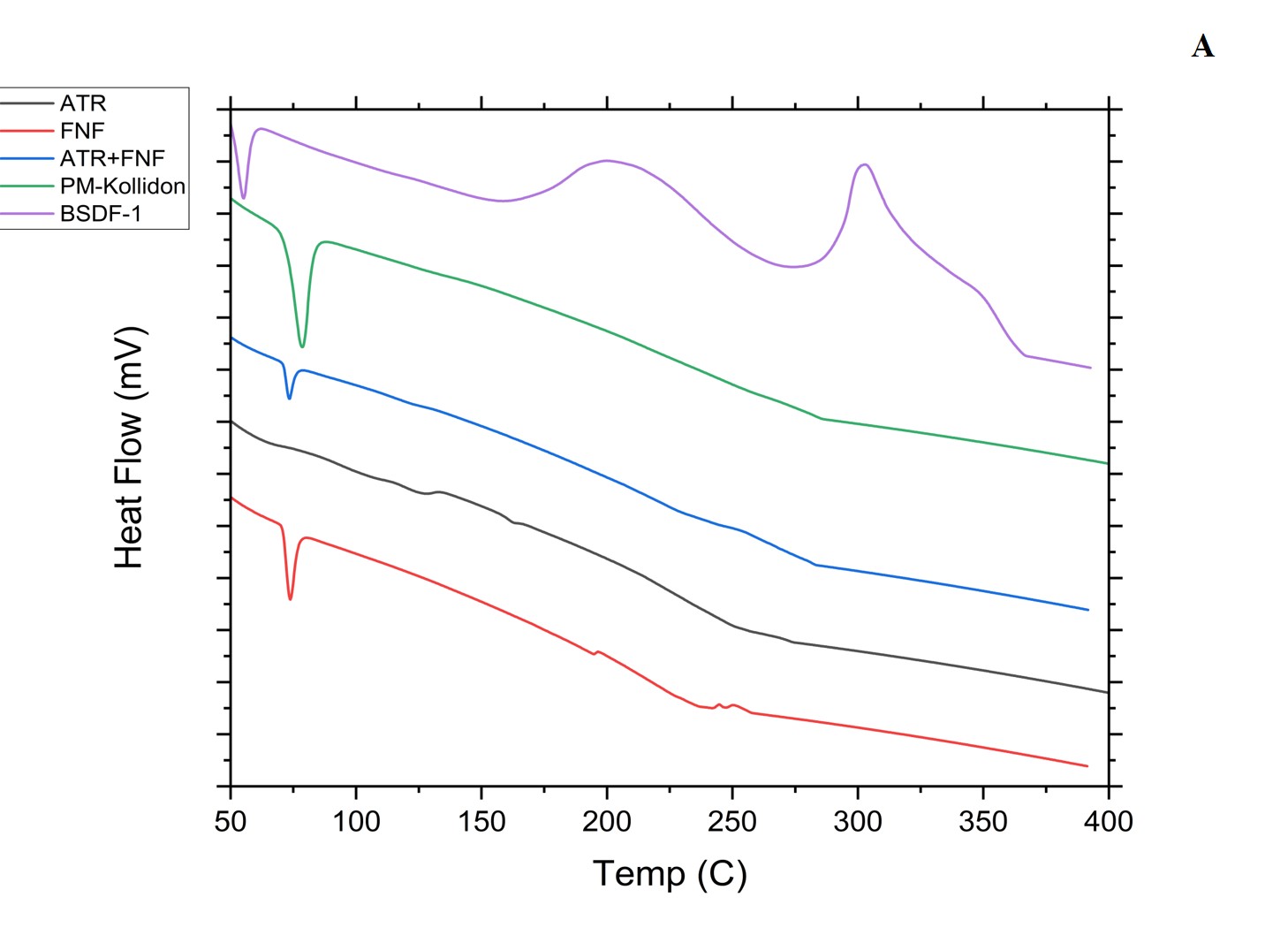
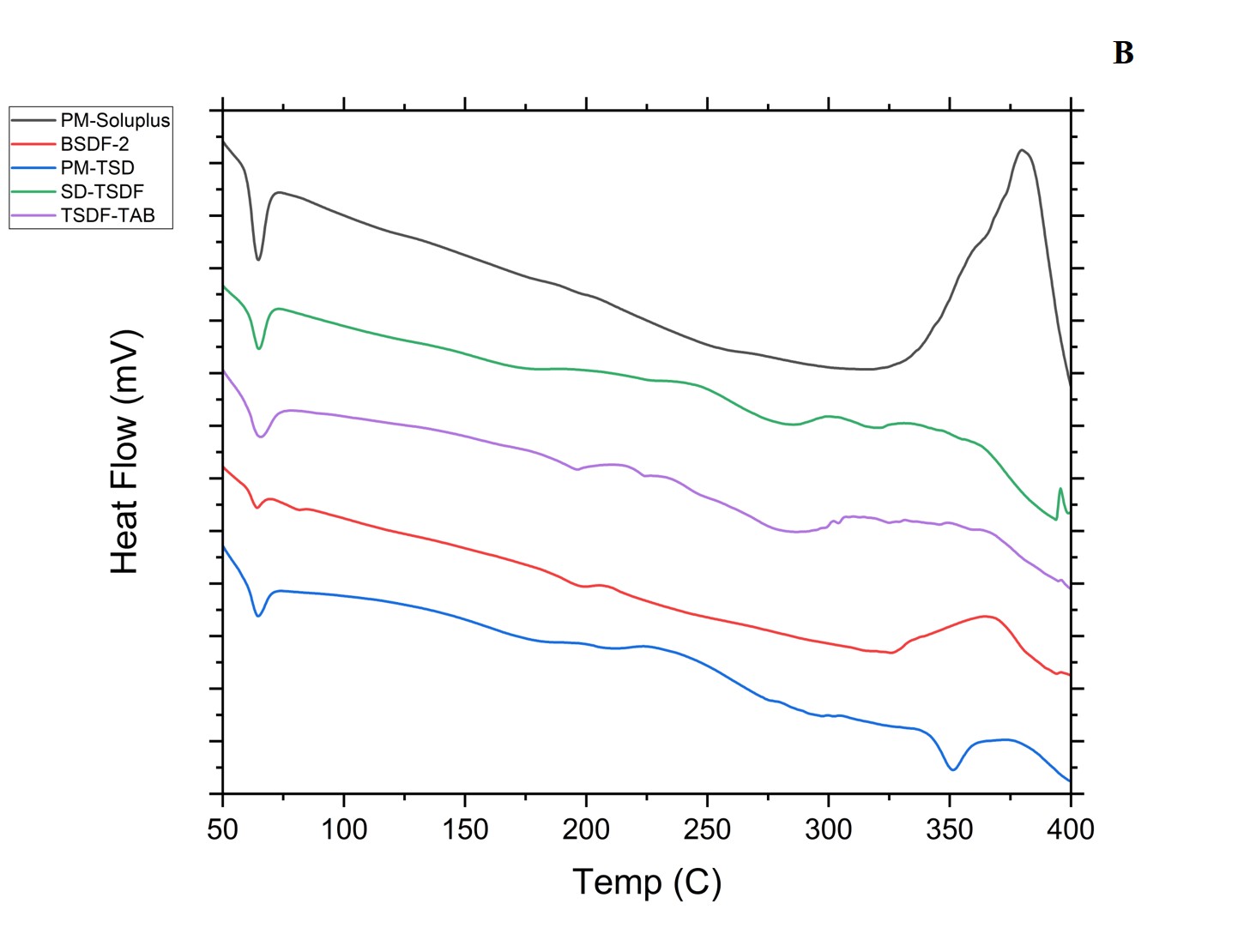
Fig. 7A and B: DSC depiction of plain drugs, polymers, physical mixture, BSD, TSD, and TSD-tablets
XRD
Another DSC pattern was observed with the pure FEN drug, confirming the presence of solid peaks at different scattered 2_theta angles of 6.03, 11.04, 11.6, 12.35, and 14.11°, while the ATR drug showed firm diffraction peaks at 2.97, 5.98, 9.01, 9.34, 10.13, 11.7, and 12.05°. Similar diffraction peaks for the studied material have also been reported previously. However, the characteristic diffraction peaks of L-NS were observed at scattering angles of 2 theta 2.9, 5.96, 6.15, 8.95, 9.29, and 10.07, which elucidated a reduction in peak intensity compared to the pure drugs, confirming the involvement of solid-state interaction at a molecular level due to the inclusion of stabilizer encasing. The XRD overlay is illustrated in fig. 8 [6].
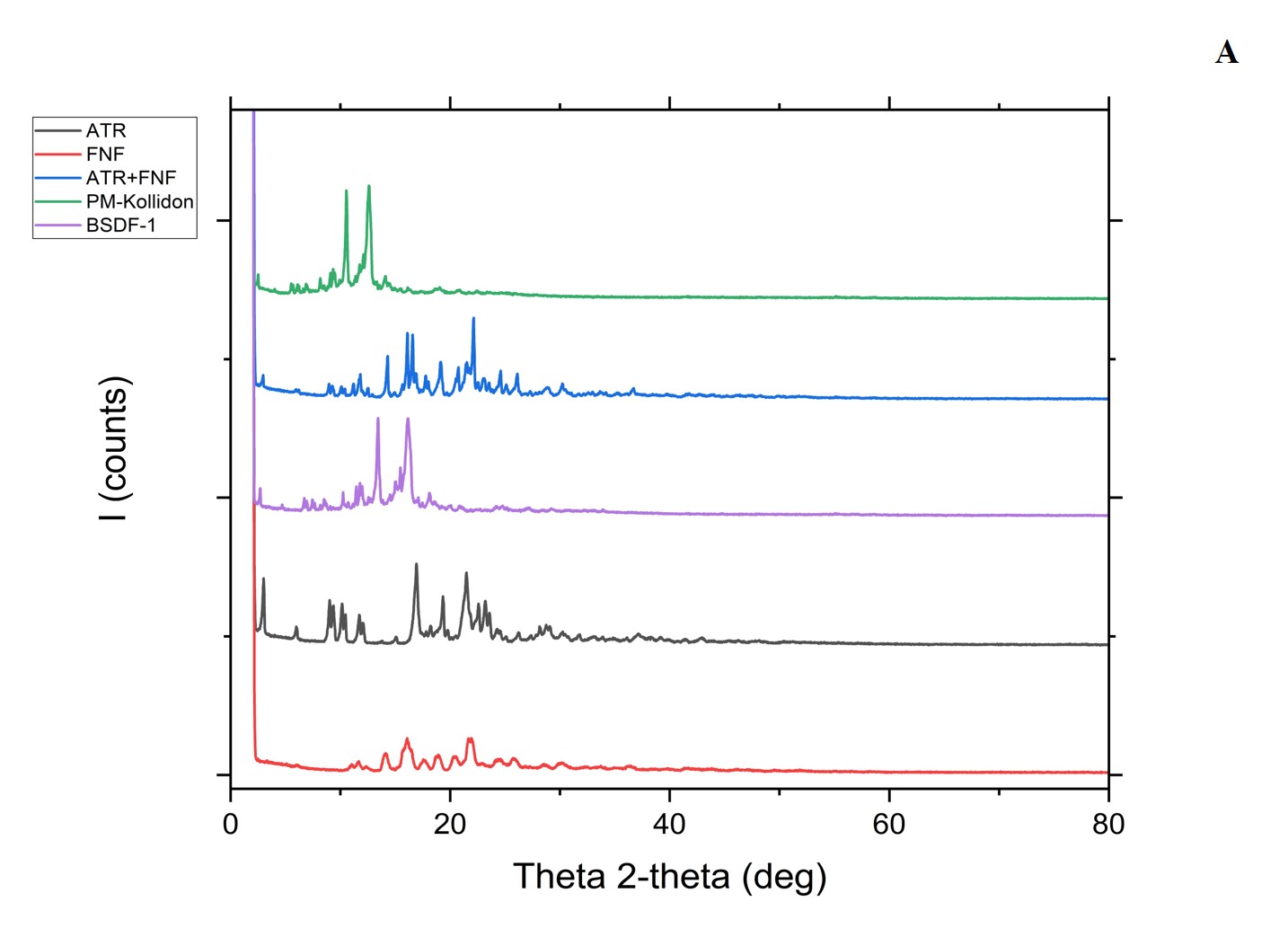
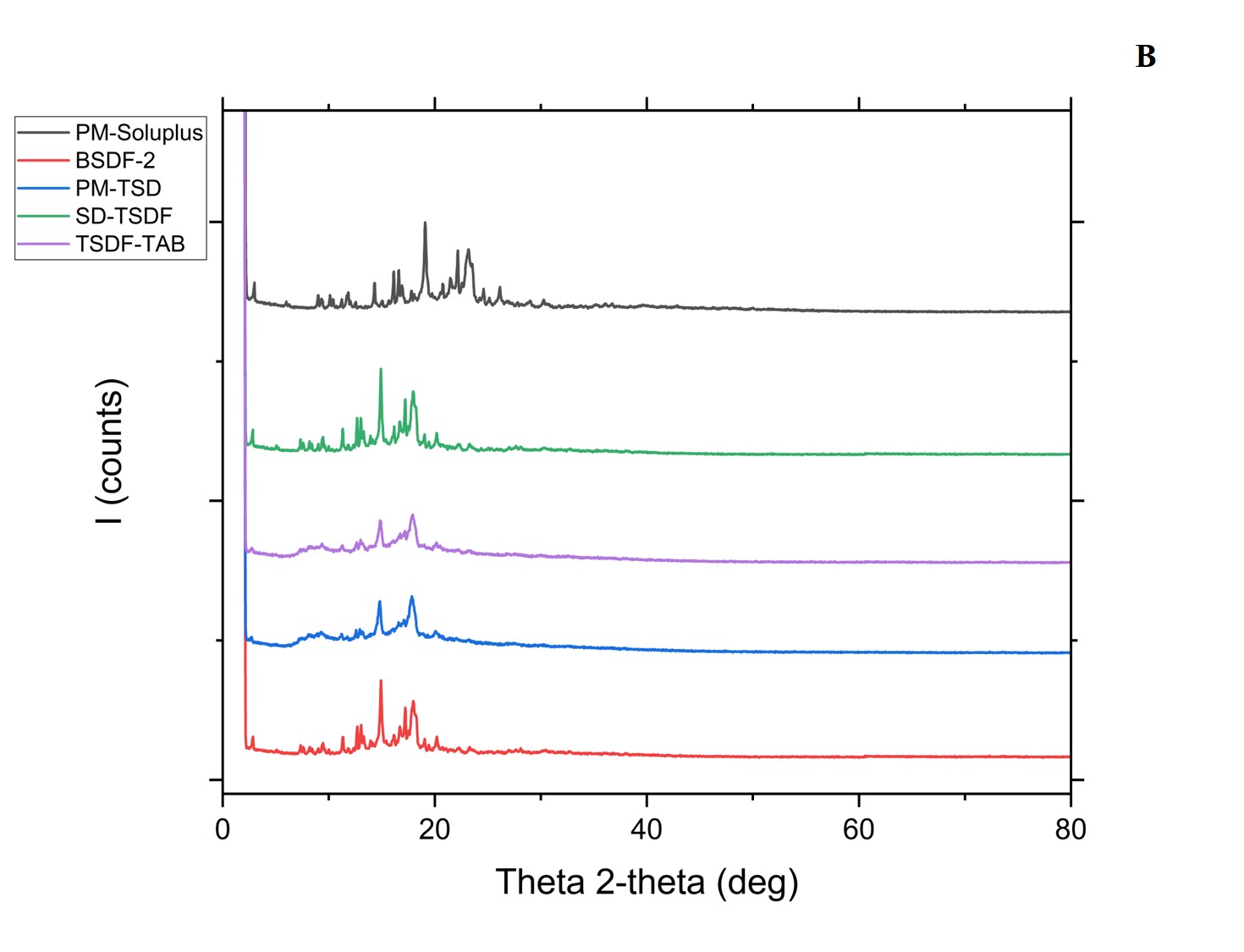
Fig. 8A and B: XRD depicting the plain drugs, polymer, physical mixture, BSD, TSD, TSD-Tablet
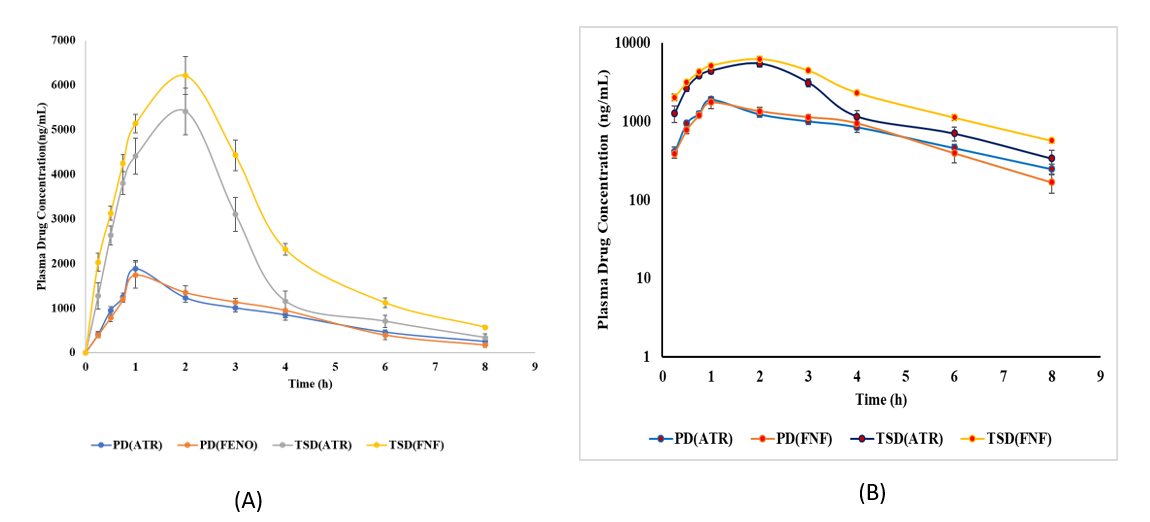
Fig. 9: In vivo release studies in animals following oral administration of plain drugs (ATR, FEN), and TSD. A) Normal Scale B) log scale. Error bars indicate SD of triplicate
Table 4: PK data for plain ATR, FEN, and TSD
| Parameters | Plain drug | Nanosuspension formulation | ||
| ATR | FEN | TSD (ATR) | TSD(FEN) | |
| Cmax (ng/ml) | 1883.64±242.93 | 1742.49± 229.91 | 5410.62± 466.75 | 6216.19± 404.19 |
| Tmax (h) | 1 | 1 | 2 | 2 |
| Half-life (h) | 2.26±0.61 | 1.60±0.34 | 2.25± 0.80 | 1.97± 0.33 |
| AUC0-t (ng. h/ml) | 6495.51±680.98 | 6532.311 ±186.34 | 16665.3± 450.82 | 22493.1± 590.17 |
| AUC0-∞ (ng. h/ml) | 7304.978±566.2 | 6918.7604±385.52 | 17766.48112± 531.20 | 24118.891±502.17 |
| MRT(h) | 3.92±01.21 | 3.36± 00.87 | 3.06±0.86 | 3.30 ±0.94 |
| Ke | 0.306 | 0.433 | 0.3070 | 0.350 |
Value expressed as mean±SD, n=3
HPLC method
Exploration of pharmacokinetic parameters
Pharmacokinetic studies
The plasma concentration versus time curve after an oral administration of the optimized nanosuspension and a commercial PD solution (0.25% w/v HPMC) is shown in fig. 9. The related pharmacokinetic information is given in table 4.
The optimised TSD markedly improved the systemic exposure of both drugs. The Cmax of atorvastatin increased from 1883.64 ± 242.93 ng/ml (plain drug) to 5410.62 ± 466.75 ng/ml (TSD), indicating a 2.87-fold enhancement. The AUC₀–t increased from 6495.51 ± 680.98 ng·h/ml to 16665.30 ± 450.82 ng·h/ml, while AUC₀–∞ rose from 7304.98 ± 566.2 ng·h/ml to 17766.48 ± 531.20 ng·h/ml, resulting in an AUC₀–∞/AUC₀–t ratio of 1.07, which suggests minimal extrapolated exposure and linear pharmacokinetics. The Cmax of fenofibrate increased from 1742.49 ± 229.91 ng/ml to 6216.19 ± 404.19 ng/ml (TSD), indicating a 3.56-fold enhancement. The AUC₀–t increased from 6532.31 ± 186.34 ng·h/ml to 22493.10 ± 590.17 ng·h/ml, while AUC₀–∞ rose from 6918.76 ± 385.52 ng·h/ml to 24118.89 ± 502.17 ng·h/ml, resulting in an AUC₀–∞/AUC₀–t ratio of 1.07, indicating linearity. A significant observation was the Tmax shift for both drugs. ATR Tmax increased from 1 hour for the plain formulation to 2 h for the TSD formulation. The FEN Tmax was extended from 1 hour to 2 h. The observed delay in Tmax indicates a polymer-mediated sustained release from the TSD matrix, likely attributable to the hydrophilic properties of Soluplus and PVP, which impede the initial drug release and absorption. The half-life (t₁/₂) and mean residence time (MRT) showed minimal variation across formulations, suggesting that the increased bioavailability resulted mainly from enhanced solubilisation and absorption rather than changes in elimination kinetics. The TSD formulation markedly enhanced the oral bioavailability of atorvastatin and fenofibrate, thereby confirming the efficacy of TSD as a delivery system [22].
Stability studies
The stability assessment conducted over a three-month period indicated that both SF8 (BSD) and KDF11 (TSD) preserved adequate drug content and kept particle size within permissible ranges at both refrigerated (4 °C) and room temperature (25 °C) conditions. The initial ATR and FEN contents for SF8 were 91.23±3.66% and 90.81±2.30%, respectively. After three months, refrigerated samples demonstrated a minor decrease (<1.5%), whereas those stored at 25 °C exhibited a more significant reduction (~3% for ATR and ~3.5% for FEN), indicating increased degradation under ambient conditions. The initial contents of ATR and FEN in KDF11 were 93.56±5.74% and 94.35±5.72%, respectively, and both remained above 92% after three months at 25 °C, indicating enhanced chemical stability in the ternary system.
TSD exhibited enhanced physical stability regarding vesicle size, with a mere 0.9% increase at 4 °C and 3.2% at 25 °C. In contrast, BSD displayed a 1.5% growth at 4 °C and a significant 9.5% increase at 25 °C. The enhanced stability of TSD can be attributed to the stronger drug–polymer interactions between Kollidon and PVP, which decrease molecular mobility and prevent aggregation, while also offering steric stabilization. The increased size of BSD at 25 °C indicates a higher likelihood of particle agglomeration and potential partial recrystallization over time.
The KDF11 TSD formulation demonstrated superior chemical and physical stability compared to SF8 BSD, consistent with its enhanced dissolution rate and in vivo bioavailability noted in previous study phases. The findings suggest that TSD is a more effective and storage-efficient method for improving the bioavailability of poorly soluble drugs such as atorvastatin and fenofibrate.
Table 5: Stability studies
| Formulation | Temp (C) | Months | ATR Content (%)±SD | FEN Content (%)±SD | Vesicle size (nm)±SD |
| SF8 (BSD) | 4 °C | 0 | 91.23±3.66 | 90.81±2.30 | 182.4±3.2 |
| 3 | 90.12±3.54 | 89.78±2.28 | 185.1±3.5 | ||
| SF8 (BSD) | 25 °C | 0 | 91.23±3.66 | 90.81±2.30 | 182.4±3.2 |
| 3 | 88.54±3.60 | 87.65±2.35 | 199.7±4.8 | ||
| KDF11 (TSD) | 4 °C | 0 | 93.56±5.74 | 94.35±5.72 | 165.6±2.9 |
| 3 | 93.02±5.68 | 93.89±5.70 | 167.1±3.1 | ||
| KDF11 (TSD) | 25 °C | 0 | 93.56±5.74 | 94.35±5.72 | 165.6±2.9 |
| 3 | 92.10±5.65 | 92.84±5.66 | 170.9±3.3 |
Value expressed as mean±SD, n=3
This study shows that TSD markedly enhances the solubility and oral bioavailability of ATR and FEN in comparison to BSD and conventional drug formulations. The enhanced performance of TSD can be explained by the synergistic interaction of Soluplus, Kollidon, and PVP. Kollidon (copovidone) facilitates micellization, enhancing drug dispersion at the molecular level. PVP improves wettability and physical stability of the formulation, preventing recrystallization and supporting sustained supersaturation. The dual mechanism improved the dissolution efficiency of the TSD system.
The results align with previous research, including Liu et al. (2013), which identified comparable synergistic effects of Soluplus and Kollidon in ternary systems, indicating enhanced amorphous stabilization and dissolution improvement. The optimized TSD formulation demonstrated an in vitro dissolution rate of nearly 99%, increasing Cmax and AUC₀–t for FEN relative to the plain drug suspension. The correlation between in vitro and in vivo data highlights the predictive validity of dissolution testing in assessing bioavailability enhancements. The observed delay in Tmax for both ATR and FEN in the TSD group suggests polymer-mediated sustained release, potentially resulting from extended retention in the gastrointestinal tract and reduced diffusion from the polymer matrix. This behavior may also influence first-pass metabolism, although further studies are required to evaluate metabolic fate and tissue distribution.
The current study effectively demonstrates the formulation's efficacy in improving solubility and bioavailability; however, long-term stability data and comprehensive toxicity assessments were not included in this scope. These aspects represent significant opportunities for future research to ensure the robustness of formulations over time and to verify safety for clinical translation. Future studies will concentrate on accelerated stability testing and systematic toxicological evaluation, critical steps in progressing the TSD system toward therapeutic application.
Future perspectives
Future research may concentrate on scaling the created TSD formulations for industrial production, assuring reproducibility, stability, and cost-effectiveness at a commercial scale, based on the encouraging results of this work. Long-term stability studies following ICH recommendations must be performed to confirm the shelf-life and integrity of the amorphous form. The integration of different polymeric carriers or innovative excipients may further improve solubility and augment bioavailability. Extending the inquiry to encompass in vivo efficacy investigations, including lipid profile alteration and cardiovascular protective effects in animal models, may yield a further understanding of the therapeutic advantages. Moreover, investigating patient-centric dosage forms, such as orally disintegrating tablets or capsules with solid dispersion, can enhance adherence. The evaluation of regulatory pathways and human clinical trials will be the final steps in converting this discovery into a feasible pharmaceutical treatment for dyslipidemia and related cardiovascular diseases.
CONCLUSION
This study developed an FDC of ATR and FEN using BSD and TSD prepared through solvent evaporation. Fourteen formulations were assessed, utilising Soluplus and Kolliphor® P 407 for BSD, and Kollidon and PVP for TSD. The optimised BSD with Soluplus exhibited over 86% drug release, indicating its potential for further enhancement via TSD. The Kollidon-based system among the TSDs demonstrated over 99% drug release, leading to its selection for pharmacokinetic evaluation. Characterization studies verified the transformation of crystalline drugs into their amorphous state and demonstrated compatibility with specific polymers. Pharmacokinetic analysis demonstrated an increase in Cmax for ATR and FEN, along with enhanced AUC and a shift in Tmax, suggesting sustained release and improved bioavailability. The enhancements indicate notable clinical benefits, such as the possibility of decreased dosing frequency, reduced therapeutic doses, and fewer side effects, thereby facilitating improved patient compliance and enhanced treatment outcomes for hyperlipidemia.
Future research will encompass ICH-compliant long-term stability studies, molecular dynamics simulations to investigate drug–polymer interactions at the molecular level, and human clinical trials to assess therapeutic potential in a clinical context. The TSD approach presents a viable framework for enhancing the efficacy and patient-centeredness of fixed-dose combination therapies in oral drug delivery.
ACKNOWLEDGEMENT
The authors sincerely thank the administration and faculty of the Department of Pharmacy, Lovely Professional University, Phagwara, Punjab, India, for providing the necessary support and facilities for their research. They also extend their heartfelt gratitude to the faculty for their continuous encouragement and guidance throughout their academic journey.
ABBREVIATIONS
ATR-Atorvastatin, FEN-Fenofibrate, BSD-Binary Solid Dispersion, TSD-Ternary Solid Dispersion, DSC-Differential Scanning Calorimetry, FESEM-Field Emission Scanning Electron Microscopy, XRD-X-ray Diffraction, ATFT-IR-Attenuated Total Reflection Fourier Transform Infrared Spectroscopy, PDI-Polydispersity Index, API-Active Pharmaceutical Ingredients, FDC-fixed-dose combinations, LDL-C-Low-Density Lipoprotein-Cholesterol, HDL-C-High-Density Lipoprotein-Cholesterol, PPAR-α-Peroxisome Proliferator-Activated Receptor-alpha, TG-Triglycerides, SIM-Simvastatin, BCS-Biopharmaceutical Classification System, ASD-Amorphous solid dispersions, PVP-Poly Vinyl Pyrrolidone, CAN-Acetonitrile, HPLC-High-Performance Liquid Chromatography, RPM-Revolutions Per Minute, w/v-weight/volume, g/mole-Grams per mole, h/h-hour/hours, %-Percent, C-Degree Celcius, Nm-Nanometers, min/min-Minutes, g/mg-Gram/Milligram, µg-Microgram, µg/ml-Microgram/milliliter, w/w-weight/weight, SD-Standard Deviation
AUTHORS CONTRIBUTIONS
All writers have made equal contributions
CONFLICT OF INTERESTS
REFERENCES
Wilkins CA, Hamman H, Hamman JH, Steenekamp JH. Fixed-dose combination formulations in solid oral drug therapy: advantages, limitations, and design features. Pharmaceutics. 2024;16(2):178. doi: 10.3390/pharmaceutics16020178, PMID 38399239.
Harano Y, Yasui K, Toyama T, Nakajima T, Mitsuyoshi H, Mimani M. Fenofibrate, a peroxisome proliferator-activated receptor α agonist, reduces hepatic steatosis and lipid peroxidation in fatty liver Shionogi mice with hereditary fatty liver. Liver Int. 2006;26(5):613-20. doi: 10.1111/j.1478-3231.2006.01265.x, PMID 16762007.
Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98(19):2088-93. doi: 10.1161/01.cir.98.19.2088, PMID 9808609.
Fiévet C, Staels B. Combination therapy of statins and fibrates in the management of cardiovascular risk. Curr Opin Lipidol. 2009a;20(6):505-11. doi: 10.1097/MOL.0b013e328332e9ef, PMID 19829109.
Górniak A, Czapor-Irzabek H, Złocińska A, Gawin Mikołajewicz AG, Karolewicz B. Screening of fenofibrate-simvastatin solid dispersions in the development of fixed-dose formulations for the treatment of lipid disorders. Pharmaceutics. 2023;15(2):603. doi: 10.3390/pharmaceutics15020603, PMID 36839925.
Rani PS, Anusha NS, Teja CP, Shakthi PV. Enhancement of bioavailability of atorvastatin and fenofibrate combination using solid dispersion technique. Int J Pharm Sci Res. 2014;5:3713. doi: 10.13040/IJPSR.0975-8232.5(9).3713-25.
Akram A, Irfan M, Abualsunun WA, Bukhary DM, Alissa M. How to improve solubility and dissolution of irbesartan by fabricating ternary solid dispersions: optimization and in vitro characterization. Pharmaceutics. 2022a;14(11):2264. doi: 10.3390/pharmaceutics14112264, PMID 36365083.
Janssens S, Van den Mooter G. Review: physical chemistry of solid dispersions. J Pharm Pharmacol. 2009;61(12):1571-86. doi: 10.1211/jpp/61.12.0001, PMID 19958579.
Srivastava A, Khan MA, Bedi S, Bhandari U. Design, optimization, and characterization of a novel amorphous solid dispersion formulation for enhancement of solubility and dissolution of ticagrelor. Int J App Pharm. 2023;15(4):296-305. doi: 10.22159/ijap.2023v15i4.47618.
Sarkar P, Biswas Majee SB. Formulation development and in vitro characterization of ternary hydrotropic solid dispersions of aceclofenac. Asian J Pharm Clin Res. 2022 Sep;15(9):174-9. doi: 10.22159/ajpcr.2022.v15i9.45158.
Sarkar P, Das S, Majee SB. Solid dispersion tablets in improving oral bioavailability of poorly soluble drugs. Int J Curr Pharm Sci. 2022 Mar;14(2):15-20. doi: 10.22159/ijcpr.2022v14i2.1961.
Farooqui P, Gude R. Formulation development and optimisation of fast dissolving buccal films loaded with glimepiride solid dispersion with enhanced dissolution profile using central composite design. Int J Pharm Pharm Sci. 2023 Jun;15(6):35-54. doi: 10.22159/ijpps.2023v15i6.47992.
Al-Hattali WS, Samuel BA, Philip AK. Enhancing fluconazole solubility and bioavailability through solid dispersion techniques: evaluation of polyethylene glycol 6000 and sodium carboxymethylcellulose systems using fiberoptics. Int J Pharm Pharm Sci. 2024;16(12):51-9.
Hirave RV, Bendgude RD, Maniyar MG, Manish K. Spectrophotometric method for simultaneous estimation of atorvastatin calcium and fenofibrate in tablet dosage form. Int J Drug Dev Res. 2013;5:38-42.
Basha M, Salama A, Noshi SH. Soluplus® based solid dispersion as fast disintegrating tablets: a combined experimental approach for enhancing the dissolution and antiulcer efficacy of famotidine. Drug Dev Ind Pharm. 2020;46(2):253-63. doi: 10.1080/03639045.2020.1716376, PMID 31937139.
Jain N, Raghuwanshi R, Jain D. Development and validation of RP-HPLC method for simultaneous estimation of atorvastatin calcium and fenofibrate in tablet dosage forms. Indian J Pharm Sci. 2008;70(2):263-5. doi: 10.4103/0250-474X.41473, PMID 20046730.
Liu J, Cao F, Zhang C, Ping Q. Use of polymer combinations in the preparation of solid dispersions of a thermally unstable drug by hot-melt extrusion. Acta Pharmaceutica Sinica B. 2013;3(4):263-72. doi: 10.1016/j.apsb.2013.06.007.
Nair AR, Lakshman YD, Anand VS, Sree KS, Bhat K, Dengale SJ. Overview of extensively employed polymeric carriers in solid dispersion technology. AAPS PharmSciTech. 2020;21(8):309. doi: 10.1208/s12249-020-01849-z, PMID 33161493.
Albadarin AB, Potter CB, Davis MT, Iqbal J, Korde S, Pagire S et al. Development of stability-enhanced ternary solid dispersions via combinations of HPMCP and Soluplus® processed by hot melt extrusion. Int J Pharm. 2017;532(1):603-11. doi: 10.1016/j.ijpharm.2017.09.035, PMID 28923766.
Choudhary A, Rana AC, Aggarwal G, Kumar V, Zakir F. Development and characterization of an atorvastatin solid dispersion formulation using skimmed milk for improved oral bioavailability. Acta Pharmaceutica Sinica B. 2012;2(4):421-8. doi: 10.1016/j.apsb.2012.05.002.
Tipduangta P, Takieddin K, Fábián L, Belton P, Qi S. A new low melting-point polymorph of fenofibrate prepared via talc-induced heterogeneous nucleation. Cryst Growth Des. 2015;15(10):5011-20. doi: 10.1021/acs.cgd.5b00956.
Gao L, Liu G, Ma J, Wang X, Zhou L, Li X. Application of drug nanocrystal technologies on oral drug delivery of poorly soluble drugs. Pharm Res. 2013;30(2):307-24. doi: 10.1007/s11095-012-0889-z, PMID 23073665.