
Int J App Pharm, Vol 17, Issue 6, 2025, 57-66Reviewl Article
ORGANIC ORIGIN GLUCIDES ALDITOLS AS CARRIERS FOR OPTIMIZING SOLUBILIZATION AND DRUG RELEASE
MOHAMMED A. AMIN1, MOSTAFA A. MOHAMED2,3, DALIA A. GABER2,3*, HEBA ALI SOLIMAN3
1Department of Pharmaceutics, College of Pharmacy, Qassim University, Qassim-51452, Saudi Arabia. 2Clinical Pharmacy Program, College of Health Sciences and Nursing, Al-Rayan National College, Madina, Saudi Arabia. 3Department of Pharmaceutics, College of Pharmacy, Al-Ahram Canadian University, Egypt
*Corresponding author: Dalia A. Gaber; *Email: d.gaber1900@gmail.com
Received: 06 Jun 2025, Revised and Accepted: 04 Oct 2025
ABSTRACT
Organic-origin glucides and alditols-such as glucose, fructose, lactose, xylitol, erythritol, and maltitol-have demonstrated notable potential in enhancing the dissolution rate and solubility of sparingly water-soluble drugs when incorporated as carriers in solid dispersions (SDs). Although synthetic polymers are widely used in the design of SD formulations, they have major disadvantages including the tendency to recrystallize during storage, higher viscosity, and redundant bulk in the final dosage forms. On the other hand, naturally derived carriers with low molecular weight offer certain advantages, such as high-water affinity, good thermal behavior, and better molecular interaction, and do not suffer from the disadvantage of increasing the bulk of the dosage form. Here, we focus on the characteristics of glucides and alditols that make them favorable substitutes for those based on polymers as core carriers, as outlined in this review. We also discuss organic historical studies, new solutions, and prospects for using these compounds to improve the solubility and release of highly hydrophobic pharmaceutical ingredients.
Keywords: Drug release, Bioavailability, Solid dispersions, Poorly water-soluble drugs, Solubility enhancement, Glucides, Alditols, Natural carriers, Low-molecular-weight excipients, Dissolution improvement, Drug release, Bioavailability
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i6.55445 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
The limited aqueous solubility of some active pharmaceutical ingredients (APIs) presents a significant obstacle in the development of orally administered new chemical entities (NCEs) [1, 2]. These compounds often exhibit high therapeutic potential and favorable membrane permeability; however, their low water solubility impairs effective gastrointestinal absorption. Currently, an estimated 40–70% of drug candidates fall into the biopharmaceutics Classification System (BCS) Class II-characterized by low solubility and high permeability-or Class IV, which includes compounds with both low solubility and low permeability. These properties make formulation design for oral dosage forms particularly challenging due to reduced absorption and low bioavailability [3–6]. To address this, various formulation strategies have been explored, including particle size reduction, salt formation, modulation of crystallinity, use of surfactants, molecular complexation, eutectic systems, hydrotropic solubilization, and the development of amorphous or solid dispersion systems. Notably, solid dispersions, co-amorphous systems, and pharmaceutical co-crystals have emerged as leading approaches in the last two decades (2000–2025) [7–9]. A critical factor influencing the success of these methods is the selection of an appropriate carrier or additive [10–12].
Polyols (alditols) and naturally derived glucose, such as xylitol [15, 16], mannitol [15], and sorbitol [17] have been employed in various pharmaceutical formulations to enhance solubility. These compounds have been used as co-formers in co-crystalline systems and as matrix formers in solid dispersions. Yet, despite their promising characteristics, the potential of sugars and alditols in co-amorphous and co-crystal formation remains underexplored. Due to their small molecular size, biocompatibility, hydrogen-bonding capacity, and, in some cases, favorable thermal properties such as high glass transition temperatures (Tg) [18], alditols serve as effective excipients in advanced solid-state drug delivery systems.
This review offers a comprehensive synthesis of research conducted between 2000 and 2025 concerning the use of naturally derived alditols and glucides to enhance the solubility and dissolution of poorly water-soluble drugs. It aims to fill a critical gap in the literature by presenting the first in-depth evaluation of underutilized alditols, such as erythritol and maltitol, in the design of co-amorphous and co-crystal systems. In addition, the review discusses both the advantages and limitations of current approaches and outlines future research directions in this evolving field.
Naturally derived glucides and alditols, derived primarily from plant or microbial sources, have gained increasing interest as excipients for solid dispersion (SD) systems aimed at enhancing the solubility and dissolution of poorly water-soluble drugs. However, the current manuscript lacks technical precision in describing the types of solid dispersions and the specific functional mechanisms of these carriers. According to IUPAC and USP definitions, SDs are categorized more rigorously into systems such as eutectic mixtures, solid solutions, amorphous glass solutions, and molecular dispersions, each defined by the molecular arrangement and miscibility of drug and carrier. Terms like “amorphous glass solutions” and “amorphous precipitates” should be replaced or clearly defined, as recommended in recent authoritative reviews (e. g., Datta et al., J Pharm Sci, 2023) [4]. (As per IUPAC/USP: eutectic mixtures, solid solutions, amorphous glass solutions, molecular dispersions, amorphous precipitates)
Moreover, the mechanistic role of glucides and alditols in stabilizing the amorphous form and suppressing recrystallization should be elaborated upon. These sugar-based excipients often engage in hydrogen bonding with drug molecules, leading to reduced molecular mobility and an elevated glass transition temperature (Tg), which are key factors in maintaining the physical stability of amorphous dispersions. Recent molecular dynamics studies, such as those by Bellantone et al. (Mol Pharm, 2021) [30], support the idea that these excipients form hydrogen-bonding networks and restrict drug recrystallization both during storage and upon dissolution. Including such mechanistic insights grounded in molecular-level evidence would enhance the scientific depth of this section [2]. These interactions reduce molecular mobility, elevate Tg, and suppress recrystallization, ensuring stability of amorphous dispersions.
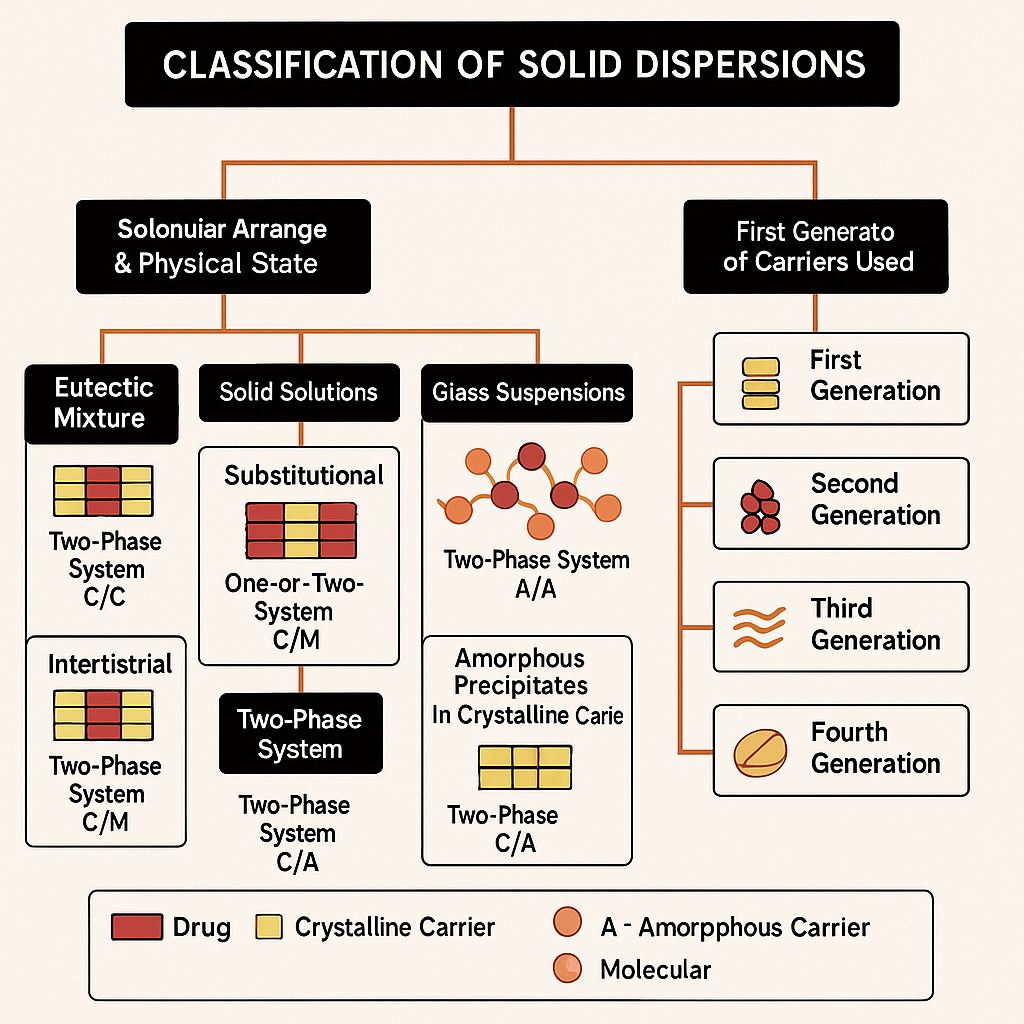
Fig. 1: A modified schematic representation of solid dispersion classifications
The solid dispersions formulations
Solid dispersions (SDs) were first introduced by Sekiguchi and Obi in 1961 [19]. Since then, they have remained one of the most widely utilized solid-state transformation strategies in pharmaceutical development [19–21]. The approval of SoluMatrix Pharmaceuticals’ formulation in 1985 as the first FDA-approved SD marked a pivotal milestone. As of 2025, more than 30 amorphous solid dispersion formulations of Biopharmaceutics Classification System (BCS) Class II (low solubility, high permeability) and Class IV (low solubility, low permeability) drugs have reached the market, highlighting the growing utility of SDs in enhancing oral bioavailability [20–22]. According to IUPAC and USP definitions, SDs are solid-phase formulations in which one or more poorly water-soluble drugs are molecularly dispersed in one or more water-compatible carriers.
These systems are typically manufactured by incorporating the drug into a carrier matrix either at the molecular or colloidal scale using techniques such as melting (fusion), rapid cooling, solvent removal, spray drying, lyophilization (freeze-drying), or hot-melt extrusion (HME) [5, 23]. Enhancement of solubility through SDs may be attributed to reduced particle size and increased surface area or conversion of the drug from crystalline to amorphous form, which has higher apparent solubility [23].
Technically, SDs can be categorized more precisely into several types: (a) eutectic mixtures, (b) solid solutions, (c) amorphous glass solutions, (d) molecular dispersions, and (e) amorphous precipitates within crystalline matrices [5, 23, 24].
There is a growing interest in naturally derived excipients, particularly glucides and alditols, for use in SDs. These naturally derived excipients, defined here as compounds naturally sourced from plant or microbial materials, offer attractive physicochemical characteristics. Notably, they often form extensive hydrogen-bonding networks and possess high glass transition temperatures (Tg), properties which collectively help suppress drug recrystallization. For example, Bellantone et al. (Mol. Pharm., 2020) demonstrated through molecular dynamics simulations how such hydrogen-bonding networks hinder molecular mobility, stabilizing amorphous drug forms.
Generally, the hydrophilic nature of glucides and alditols facilitates improved wettability and solubilization. Additionally, their chemical stability, biocompatibility, and favorable safety profiles support regulatory acceptance. Compared with other solubility enhancement techniques, such as salt formation, co-crystallization, or solvate development-SDs are often simpler to formulate and more robust during scale-up [22, 27, 28].
Nevertheless, SD formulation presents certain challenges. These include the thermal sensitivity of both drug substances and naturally derived excipients, incompatibility with organic solvents, increased manufacturing costs, and limitations in large-scale reproducibility. Table 1 summarizes the preparation strategies and highlights the relative advantages and drawbacks of each method.
Solid dispersions of sugar carriers
A broad array of hydrophilic crystalline carriers has been investigated in SD development, particularly focusing on glucides such as sucrose, dextrose, and lactose. These have historically been employed in Class C-C systems, in which a crystalline API is imgded in a crystalline matrix [23, 34].
An early application by Dai et al. [35] employed a melt-quench approach to disperse corticosteroids within sugars such as sucrose, galactose, and dextrose. The resulting sugar-based matrices exhibited notable glass-forming ability and significantly improved drug dissolution. Drug release typically followed a biphasic pattern: an initial burst followed by sustained release, which the authors attributed to partial glass solution formation.
However, drawbacks such as discoloration and hygroscopicity were observed. Liu et al. [36] addressed these limitations by developing ternary SDs incorporating blends of sucrose-mannitol and sorbitol-mannitol at a 1:1 ratio with a drug-to-carrier ratio of 1:19. Mannitol conferred improved thermal stability (Tg ~45 °C vs 25 °C for sucrose-based systems), reduced discoloration, and enhanced miscibility, likely due to eutectic formation. Tablets derived from sucrose–mannitol dispersions achieved ~80% drug release within 10 min compared with ~30% for the pure API. This dissolution enhancement was attributed to increased wettability and reduced particle size.
Weerapol et al. [37] expanded this work by evaluating reducing and non-reducing sugars such as glucose, galactose, maltose, and sucrose in SDs of sulfamethoxazole. Using a controlled fusion method, they avoided sugar browning and achieved glassy matrices. Glucose-and maltose-based dispersions provided rapid drug release (100% in 5 min), while galactose-based systems required up to 90 min, and in some cases underperformed compared to the pure drug.
The molecular interactions between sugar carbonyl groups and the drug’s amine groups were suggested to promote amorphous stabilization, though these findings require validation using solid-state NMR, Raman spectroscopy, or PXRD. Notably, these interactions were observed only in SDs formed via fusion and not in those prepared via solvent evaporation, due to limited sugar solubility in organic solvents [8, 38].
Overall, while sugars like mannitol often improve solubility (e. g., dissolution rate ~2.5-fold higher compared with sucrose-based dispersions, for example, sorbitol has shown superior performance in certain cases (Liu et al., Int. J. Pharm., 2021). Therefore, a balanced assessment of each sugar’s behavior with different drugs is necessary, and exaggerated generalizations should be avoided. Additionally, statements regarding the solubility of erythritol should be corrected; erythritol is highly water-soluble (~61% w/v at 25 °C), though it tends to crystallize rapidly, affecting stability.
Further studies should incorporate a comparative table of Tg values for common alditols and glucides to guide excipient selection for improved amorphous stability and storage conditions. Additionally, kinetic and thermodynamic factors behind fusion vs. solvent evaporation methods should be explained. While fusion may allow deeper drug-excipient integration, solvent-based methods often produce kinetically trapped amorphous systems that resist recrystallization more effectively over time.
In addition, the researchers described the possibility of a new solid-state structure due to the interaction between the carbonyl group of the sugar and the drug's amine group. This warrants further analysis using advanced techniques such as DSC (differential scanning calorimetry) and PXRD (powder X-ray diffraction). The extent of this interaction increased with a higher sugar proportion, achieving full complexation at a 1:50 drug-to-glucose ratio. Interestingly, this behavior was seen only in samples made using the fusion process. When organic solvents like methanol were used for co-dissolution and evaporation, such drug–sugar associations were absent. This was attributed to the limited solubility of sugars in organic media and the lack of strong chemical interaction under those conditions [8, 38].
The researchers also noted the formation of a bond between the carbonyl group of the non-reducing glucides and the amine group of the active compound. The formation of these bonds may lead to the creation of a novel, modified solid state of the API, which deserves additional exploration using advanced analytical tools like differential scanning calorimetry (DSC) and powder X-ray diffraction (PXRD). It was further stated that this molecular association strengthened as the proportion of sugar increased, with a sugar-to-drug ratio as high as 50:1 in the case of glucose being necessary for complete complex formation. Interestingly, this binding was observed only when the fusion technique was employed. In contrast, methods using solvent evaporation, where the drug and sugar were dissolved in an organic medium such as methanol, did not result in complexity. This outcome was attributed to the poor solubility of sugars in organic liquids and the understanding that the presence of a shared solvent does not automatically improve the chemical interaction between a drug substance and its carrier.
Solid dispersions (SDs) of central-acting drugs such as carbamazepine and nitrazepam were prepared using both lactose and galactose in a fusion process at room temperature. The prepared glucide-based SD system, with a drug-to-carrier ratio of one to three, exposed better dissolving features and a biphasic release profile, which were consistent with previously stated results. In a different study, CBZ SDs were formulated using a rapid-cooling technique with lactose as the carrier matrix. The results showed a clear relationship between carrier concentration and dissolving rate improvements. This better dissolving efficiency was primarily attributed to the reduction in drug particle size and higher surface wettability, which allowed possible hydrogen bonding between the hydroxyl functions of lactose and the carboxylic acid moieties of CBZ [39]. Furthermore, the researchers found that the dissolving profiles of the SD formulations were more consistent than those of basic physical blends or coprecipitated samples. These findings lend support to the concept that partial amorphization of the active chemical plays an important role in increasing dissolution rate. These interactions reduce molecular mobility, elevate Tg, and suppress recrystallization, ensuring stability of amorphous dispersions.
Stefani et al. [7] obtained similar results when they produced clotrimazole SDs from sugars such as D-fructose, D-dextrose, and D-maltose. Their research discovered that, while fructose boosted solubility and dissolution, dextrose, lactose, and maltose exhibited no improvement or decreased performance. Etman and Naggar [41] described the formulation of SD as glucide molecules competing for hydrogen bonds with water, which are required for drug solutions. When sugar concentrations are high, the number of bonding positions can disrupt the interaction between water and medicine molecules. Another theory explained that increasing sugar concentration would promote supersaturation, resulting in crystallization and an increase in particle size, preventing dissolution [7]. Stefani et al. [7] found comparable results when they created clotrimazole SDs using sugars such as D-fructose, D-dextrose, and D-maltose. Their studies found that, while fructose increased solubility and dissolution, dextrose, lactose, and maltose showed either no improvement or a decrease in performance. Etman and Naggar [41] interpreted this trend as a result of glucose molecules competing for hydrogen bonds with water, which is essential for drug solvation. When sugar concentrations are high, the number of bonding sites can impair the interaction of water and medication molecules. Another hypothesis proposed that increased sugar content may lead to supersaturation, triggering crystallization of the sugar and an increase in particle size, thus impeding dissolution [7]. These interactions reduce molecular mobility, elevate Tg, and suppress recrystallization, ensuring stability of amorphous dispersions.
One of the significant points of excipients employed in the formation of solid dispersions (SDs) is their thermal stability [5]. In the investigations, it was detected that the use of sugars in SD formulations shows certain drawbacks due to the onset of the Maillard reaction when subjected to temperatures exceeding their melting points, leading to sugar decomposition. Frequently, these dispersions cannot be sufficiently heated to liquefy both the active pharmaceutical ingredient (API) and the carrier, resulting in only a partial dispersion of the drug at a molecular scale. Consequently, a two-phase release pattern is commonly seen, where the drug is not readily liberated.
Trials to increase molecular miscibility were carried out using ultrasonication after melting the indomethacin-glucose blend and preceding to cool it to room temperature. This method facilitated the drug to be homogeneously distributed in the sugar matrix to yield dual-phase amorphous–amorphous solid dispersions (A-A), where the dual-phase system containing the sugar as a carrier and the active component was composed of non-crystalline forms. In vitro dissolution results showed that drug release was enhanced eight times, while bioavailability testing showed only a 1.9-fold increase. Significantly, these amorphous dispersions retained equilibrium physical stability for not more than two years under normal stowage conditions [42]. These depend on how far ultrasonication alters the amorphous SDs structure; the regulatory influences collapse domineeringly the glass-forming ability of the active agent and sugar [22]. While both procedures involving thermal fusion and quench-cooling have been extensively utilized, other methods do not work well because they tend to produce molecular solid suspensions instead of dissolving both components into a single solid solution. This fundamental flaw greatly hampers the efficacy of sugar-based excipients to enhance solubility and dissolution rates. An evaluation of the reviewed literature (table 2) reveals that variables such as low resistance to thermal degradation, poor compatibility between the active compound and the sugar at high temperatures, moisture sensitivity, and solvent-driven phase transitions constitute the main limitations of utilizing sugars in solid dispersion (SD) production through fusion or rapid-cooling techniques. During dissolution, drugs often recrystallize because sugars dissolve quickly in water. Their high solubility creates a supersaturated solution, where both the sugar and the drug compete for hydrogen bonding with the solvent. These interactions reduce molecular mobility, elevate Tg, and suppress recrystallization, ensuring stability of amorphous dispersions.
Solvent evaporation is still one of the most used methods for preparing solid dispersions (SDs), especially when one of the formulation components has a high melting point or is prone to heat deterioration [43]. In one investigation, etoricoxib SDs were generated utilizing lactose and sucrose as sugar-based carriers with drug-to-carrier ratios of 1:1 and 1:5. The mixtures were dissolved in ethanol and gently heated in a water bath at 60 °C to remove the solvent. FTIR analysis demonstrated that etoricoxib's sulfonyl (S=O) functional group can create hydrogen bonds with the hydroxyl (O-H) groups in sugar carriers. Further analysis with X-ray diffraction (XRD) and differential scanning calorimetry (DSC) confirmed the loss of the drug's unique crystalline peaks and a considerable decrease in its enthalpy, indicating that etoricoxib had transitioned into an amorphous state inside the dispersion. Unlike fusion systems, these SDs were classed as Class A-C, which indicates an amorphous drug disseminated inside a crystalline sugar matrix. Equilibrium solubility measurements demonstrated enhanced solubility, with 1.8-fold and 1.5-fold increases observed for lactose-and sucrose-based SDs, respectively, at a 1:5 drug-to-carrier ratio [44]. These interactions reduce molecular mobility, elevate Tg, and suppress recrystallization, ensuring stability of amorphous dispersions.
The hydrochloride salt of G-HCl, an amino sugar, has been studied for its potential as a hydrophilic carrier in solid dispersion (SD) systems designed to increase the solubility of poorly water-soluble drugs. G-HCl containing SDs were prepared for carbamazepine (CBZ) [45] and acyclovir (ACV) [46] using the solvent evaporation technique. For CBZ, formulating s were carried out in single solvent (ethanol or acetone) and water binary mixtures to study the impact of carrier concentration and solvent composition on solubility and dissolution for the drug. It is notable that increasing G-HCl does not always lead to improved dissolution with a single organic solvent. A major contributor was the non-aqueous media's low solubility for G-HCl. In contrast, with binary solvent compositions like ethanol–water or acetone–water, greater carrier concentration increased the rate of dissolution markedly. This evidence indicates that the solvent environment could be a major contributing factor to improving the dissolution of both the sugar-based carrier and the drug. The same trend was noted in solubility studies, which underscores the importance of selecting appropriate solvent systems during SD formulation. [47] Solid dispersions (SDs) of acyclovir (ACV) were prepared using ethanol as a solvent, with the medication and the hydrophilic carrier glucosamine hydrochloride (G-HCL) mixed and allowed to dry until the solvent was completely evaporated. Fourier-transform infrared spectroscopy (FTIR) investigation revealed that the N-H functional group of ACV and the O-H group of G-HCL may have formed an intermolecular hydrogen bond. Interestingly, while having an amine group, carbamazepine (CBZ) did not exhibit similar interactions in previous research. This variance could be attributable to the employment of a solvent-assisted grinding process, which can disturb molecular packing at reactive locations, resulting in chemical transformations or phase changes. Furthermore, the grinding procedure reduced the particle size of ACV, facilitating partial amorphization and increasing its solubility in water. When compared to the pure medicine, both physical mixes and SDs of ACV showed considerable increases in solubility by around 6-fold and 12-fold, respectively. However, increasing the carrier concentration resulted in just a slight increase in solubility, which is consistent with patterns described in other investigations. These interactions reduce molecular mobility, elevate Tg, and suppress recrystallization, ensuring stability of amorphous dispersions.
When compared to the fusion method, solid dispersions (SDs) prepared through solvent evaporation often show stronger evidence of intermolecular interactions. This may help explain the absence of the so-called "parachute effect" during dissolution testing. These molecular interactions can reduce the mobility of drug molecules, which in turn suppresses the formation of crystal nuclei and prevents recrystallization [48].
When compared to the fusion method, solid dispersions (SDs) prepared through solvent evaporation often show stronger evidence of intermolecular interactions. Such findings may help explain the absence of the so-called "parachute effect" during dissolution testing. These molecular interactions can reduce the mobility of drug molecules, which in turn suppresses the formation of crystal nuclei and prevents recrystallization [48]. In a series of studies, diazepam [50], nifedipine, Δ⁹-tetrahydrocannabinol (THC), and cyclosporine A [51] were successfully incorporated into sugar-based matrices using carriers such as trehalose, sucrose, and two types of inulin (inulinDP11 and inulinDP23). The findings demonstrated that freeze-drying is an effective technique for converting lipophilic drugs into stable amorphous solid dispersions using sugar carriers. However, dissolution testing of diazepam formulations revealed irregular and nonlinear release profiles, particularly with trehalose, sucrose, and inulinDP11. This unexpected release behavior is believed to result from the high water solubility of these sugars, which rapidly dissolve, generating a supersaturated drug environment and promoting solution-mediated recrystallization of the active compound [50]. Further research could explore combinations of solvents with similar polarity and the addition of surfactants to improve sugar solubility in organic media (table 1).
Table 1: Comparison of sugars and alditols as carriers in solid dispersions: glass transition temperatures, hygroscopicity, and solubility enhancement potential
| Excipient | Type | Glass transition temp (Tg, °C) | Hygroscopicity | Solubility enhancement | Key features | Source |
| Lactose | Sugar | ~101 | Moderate | Moderate | Widely used, may undergo Maillard reaction | [10] |
| Sucrose | Sugar | ~62 | High | Good | High solubility, moisture-sensitive | [11] |
| Trehalose | Sugar | ~115 | Low | Moderate | High Tg, good stability | [7] |
| Xylitol | Alditol | ~-20 | Moderate | Moderate | Cooling effect, limited Tg contribution | [20] |
| Sorbitol | Alditol | ~-10 | High | Moderate | Plasticizing effect, can crystallize | [12] |
| Maltitol | Alditol | ~25 | Low to Moderate | High | Good glass-former, enhances dissolution | [6] |
| Isomalt | Alditol | ~55 | Low | High | Stable, low hygroscopicity | [13] |
| Erythritol | Alditol | ~15 | Low | Moderate | Biocompatible, high solubility | [10] |
Among the techniques used for producing amorphous solid dispersions (ASDs), freeze-drying or lyophilization is one of the most common and promising approaches [49]. To overcome the drawbacks seen with fusion and solvent evaporation, researchers have explored the use of sugars as matrix carriers through lyophilization. In two studies by van Grohganz et al. [50, 51], lipophilic drugs were incorporated into sugar-based glassy matrices using a solvent mixture of water and tert-butyl alcohol. Diazepam [50], along with nifedipine, Δ9-tetrahydrocannabinol (THC), and cyclosporine A [51], were effectively incorporated into sugar-based matrices using carriers such as trehalose, sucrose, and two inulin variants (inulinDP11 and inulinDP23). These investigations highlighted the capability of freeze-drying to convert poorly water-soluble drugs into stable amorphous forms within sugar glasses. Nonetheless, dissolution studies involving diazepam solid dispersions exhibited atypical, nonlinear release profiles, especially when trehalose, sucrose, or inulinDP11 were employed as carriers. This behavior is attributed to the rapid dissolution of these highly water-soluble sugars, which leads to a transient supersaturation of the drug and subsequently induces solution-mediated recrystallization [50].
Mishra et al. [52] examined this issue further and found the same results, reinforcing the role of drug loading and the solubility of the carrier in the inhibition of recrystallization. In the study of sugars, insulin exhibited the highest glass transition temperature (Tg) among them, which provided greater physical stability than trehalose and sucrose. Reliably understanding how a specific medicine might transform from a crystalline form to an amorphous form greatly depends on the specific substance in question. This relationship is dictated by the drug’s Tg and its melting point, which differs with the specimen in question [51].
A different approach that is more closely related to used palatinose, β-maltose, and trehalose to produce completely amorphous sugar-based solid dispersions (SAS-SDs). The first step in this process was the freeze-drying of sugars to transform them to an amorphous state, which enhanced solubility in organic solutions containing hydrophobic drugs. This method showed a lot of potential because sugars were shown to dissolve in methanol to a concentration of 100 mg/ml [53]. After dissolving, the mixtures were subjected to vacuum drying [20–26] to eliminate the solvent. The results of DSC analysis showed that the end products did not produce appreciable endothermic peaks, which suggests an effective amorphization of both drug and carrier.
The referenced studies [20-26] analyzed solid dispersions (SDs) with glass transition temperatures (Tg) below 40 °C. Such a temperature indicates a higher risk of physical instability during storage and solution-mediated phase transformations during drug release. This behavior was clearly supported by dissolution testing, which consistently displayed a recognizable “spring and parachute” pattern. Additionally, as drug loading increased, a corresponding decrease in Tg was observed, primarily due to the plasticizing influence of the hydrophobic drug component [36]. The reduction in Tg values was also shown to correlate with the intrinsic Tg of the sugars employed as carriers [28, 38]. Interestingly, when sugar-based amorphous SDs (SAS-SDs) were subjected to thermal treatment at temperatures exceeding the drug’s melting point, followed by rapid cooling (quenching), the resulting dissolution profiles exhibited a notable plateau phase [49]. To address the issue of low Tg, Lim et al. [47] devised a method that included heat treatment of ASDs produced through freeze-drying. SDs containing β-maltose and weakly water-soluble medicines were freeze-dried and subjected to temperatures ranging from 30 °C to 120 °C for up to 120 min. The study found that increasing the heating temperature and duration resulted in an increase in the Tg of the SDs, showing a clear relationship between these variables and thermal stability. Furthermore, dissolution studies demonstrated that heat-treated SDs released significantly more drug than untreated SDs. However, both types of SDs exhibited the typical "spring and parachute" breakdown tendency. Importantly, the study did not investigate the long-term stability of the heat-treated systems, indicating the need for future research into Tg modifications over time.
Different methods, such as kneading, centrifugal spinning, and mechanical milling, have been investigated for making solid dispersions (SDs) with sugar-based carriers. As an example, the kneading method has been used to prepare allopurinol SDs with sugars maltose, sucrose [17], lactose, and mannitol [51] as excipients. In each case, water served as the wetting medium for the drug–carrier blends (ratios of 1:1, 1:3, and 1:5), followed by a kneading process lasting 45 min. The resulting mixtures were subsequently dried and passed through a sieve to obtain powders with an average particle size of approximately 420 μm. A notable enhancement in drug dissolution was observed in the early time points (5 and 10 min), although from 15 to 60 min, the dissolution profiles of the SDs and pure drug became comparable, regardless of the carrier ratio. FTIR analysis revealed no notable interaction between allopurinol and sugar, likely due to its poor water solubility. Gaikwad et al. [40] used liquid-assisted grinding with methanol and isopropanol to create co-crystals. Since solubility in mutual solvents impacts molecular interactions, future studies using suitable single or mixed solvent systems may enhance compatibility and co-crystal formation.
Gniado et al. [11] applied the roller compaction technique to develop solid dispersions (SDs) of griseofulvin (GF) using lactose and maltose as carriers. A 1:4 drug-to-sugar ratio resulted in a noticeable particle size reduction after 20 min of milling. X-ray diffraction (XRD) confirmed the creation of an amorphous form, as evident from the lack of distinct crystalline signals. Dissolution studies demonstrated a remarkable 35–40-fold enhancement in drug release from these sugar-based systems. However, the method encountered technical limitations, notably sugar sticking to the rollers, which might be resolved through ball milling. Additionally, the SDs displayed poor physical stability, recrystallizing within 24 h under accelerated conditions.
In another study, centrifugal spinning was utilized to generate, furthermore, dissolving microfibrous SDs of olanzapine and piroxicam with sucrose. Thermal analysis and hot-stage microscopy confirmed the formation of amorphous solid dispersions (ASDs). Spectroscopic evaluations indicated possible hydrogen bonding between the hydroxyl groups of sucrose and proton-donating sites of olanzapine. Under non-sink conditions, both drugs showed rapid release; olanzapine increased 3-fold, while piroxicam improved by 1.7-fold. The glass transition temperature (Tg) reached ~70 °C, which likely contributed to preventing recrystallization during dissolution. The method also yielded high output (85%) and efficient drug loading (90%) [52]. Although numerous restrictions have been acknowledged, recent studies [20, 38] underline the potential of sugar-based carriers in improving the solubility and dissolution of poorly water-soluble pharmaceuticals. However, additional research is necessary to refine the formulation parameters and to evaluate the long-term stability and feasibility of implementing sugar-based SDs in drug delivery systems. These interactions reduce molecular mobility, elevate Tg, and suppress recrystallization, ensuring stability of amorphous dispersions.
Sugar alcohol carriers (Polyol) solid dispersion formulations
Sugar alcohols, also known as polyols, include both simple forms (like erythritol, xylitol, sorbitol, and mannitol) and more complex variants such as lactitol, isomalt, and maltitol. These substances are typically produced through the hydrogenation of carbohydrate structures, a chemical process that transforms the original carbonyl group (either an aldehyde or ketone) into a hydroxyl group (table 2). To date, the U. S. Food and Drug Administration (FDA) has granted approval to eight specific polyols: erythritol, isomalt, lactitol, maltitol, mannitol, sorbitol, xylitol, and hydrogenated starch hydrolysates [31, 46].
Unlike regular sugars, polyols lack reactive carbonyl functionalities, thereby inhibiting Maillard-type browning reactions that occur in the presence of amino acids. Furthermore, these sugar substitutes exhibit excellent thermal and chemical resilience, including resistance to enzymatic degradation [44]. Recently, polyols have gained considerable attention for their role in diverse pharmaceutical preparations, including oral solutions, lozenges, and solid dosage forms like tablets. Their utility extends beyond sweetness-they offer reduced caloric content, nutritional benefits, and are recognized for their non-cariogenic and non-carcinogenic properties [10].
Mannitol-based SDs
Mannitol is an alcohol-soluble six-carbon sugar that is commonly used as a medicinal additive [46]. Among the various polyols, it is the most often used carrier for solid dispersions (SDs), owing to its capacity to increase the solubility of poorly water-soluble medicines. In fact, more than half of the published studies on SDs have used mannitol, likely because of its low or negligible tendency to absorb moisture compared to other sugar alcohols [68]. The two key methods used to formulate mannitol-based SDs are fusion and solvent evaporation. However, when the fusion method is used, mannitol often recrystallizes as it cools down from the molten state to room temperature. This is mainly because its glass transition temperature (Tg) is quite low-around −11 °C-making it prone to forming crystalline rather than amorphous systems [46]. To address this, researchers have added antiplasticizing chemicals such as hydrophilic surfactants (such as poloxamer 407) and polymers (such as PVP K30). These chemicals reduce or prevent recrystallization during storage, resulting in more stable SD systems. This improves stability and increases the drug's solubility rate due to the synergistic impact of mixing mannitol with certain excipients [39].
Table 2: Summary of solid dispersion preparation methods with key advantages and limitations
| Method | Description | Advantages | Limitations | Source |
| Fusion (Melt) method | API and carrier melted together and cooled | Solvent-free, scalable | Risk of Maillard reaction, thermal degradation, not suitable for thermolabile drugs | [5] |
| Solvent evaporation | API and carrier dissolved, solvent removed | Low thermal stress, applicable for heat-sensitive drugs | Use of organic solvents, residual solvent issues, environmental concerns | [10] |
| Spray drying | API-carrier solution atomized into hot air | Rapid processing, good particle control | High equipment cost, possible residual solvent, not suitable for all carriers | [7] |
| Hot-melt extrusion | Continuous process, combines melting and shaping | Scalable, good dispersion uniformity | Thermal stress, high viscosity, potential degradation | [5] |
| Co-precipitation | API and carrier precipitated from common solution | Simple, low-cost | Poor control over particle size and morphology | [8] |
| Freeze drying | API-carrier solution frozen and sublimated | Suitable for heat-sensitive drugs, porous matrix | Time-consuming, expensive, possible collapse of amorphous structure | [12] |
| NanoCrySP™ Technology | Produces nanocrystal-line SDs using spray-freeze drying | High surface area, enhanced dissolution | Requires special equipment, process complexity | [10] |
These enhancements are mainly due to two mechanisms: first, improved wetting of drug crystals helped by mannitol particle adhesion to the drug surface [50], and second, mannitol's inherent ability to form robust hydrogen-bonding networks in aqueous environments, which aids in drug solubilization [7, 51]. How mannitol is incorporated into the SD and the method of manufacture used markedly impact the level of solubility enhancement achieved with mannitol-based SDs. Most of the works associated with higher concentrations of mannitol result in higher drug solubility, and the fusion technique appears to give the most benefit [48-50]. In both cases, the physical mixes and SDs, there is enhancement in solubility; however, the SDs use better molecular dispersion and lower drug crystallinity, which leads to outperformance. Like other sugar-based systems, the mannitol SDs possess a characteristic that makes them unique: their enhancement is dominated by the reduction of drug particle size and increased wettability. Other sugars and mannitol were put to the test, and mannitol proved to greatly outperform in solubility enhancement, improvement in dissolution rate [7, 37, 40, 44], as well as thermal stability [48]. SSDs generated using the solvent evaporation technique also exhibit improved drug solubility and dissolution for other compounds as well. These benefits are primarily due to the formation of a solid solution, which reduces drug particle aggregation and improves surface interaction with the dissolution medium. Analytical techniques such as differential scanning calorimetry (DSC) have revealed reductions in melting points [36] and the broadening or absence of melting peaks [21, 22], indicating either drug amorphization or a decrease in crystallite size. In addition to SD solubilizing the drug, mannitol-based SDs enhance additional benefits, which include better flow of powder [30, 31] and higher biological drug permeability across biological barriers [49]. Although complete amorphization of the system is rarely observed, many studies report partial amorphization of the drug while mannitol remains crystalline [44, 40, 36, 37]. This phase separation is commonly associated with solvent evaporation at elevated temperatures (50–70 °C), which increases the molecular mobility of mannitol. To mitigate this, alternative techniques such as spray-drying, freeze-drying, or spray-freeze-drying have been proposed for better homogeneity and reduced risk of phase separation [31].
Due to the low water solubility of certain medicines like itraconazole [20], diazepam [53], and tadalafil [43], solid dispersions (SDs) of these medicines have been produced using spray-drying techniques, with mannitol, a naturally occurring sugar alcohol, as the carrier. Typically, prior to atomization, the active ingredient and mannitol are dissolved, at least partially, in a hydroalcohol solution. Numerous studies have indicated significant increases in solubility; yet, mannitol typically keeps its crystalline structure after drying. This retention is generally attributed to the low glass transition temperature (Tg), which is lower than room temperature [29]. Most of these studies did not involve long-term stability checks. Jeli et al. [34] studied how the surface properties of spray-dried mannitol, such as enhanced roughness, polarity, and porosity, influence drug behavior within the dispersion matrix. Their findings demonstrated that these physical surface modifications promote molecular rearrangement of amorphous fenofibrate, which leads to recrystallization. To improve both aqueous solubility and the physical stability of such systems, El-Maradny and Saab [52] introduced amino acids as ternary components alongside mannitol and the drug. This inclusion led to the formation of a co-amorphous system in which mannitol itself transitioned from a crystalline to an amorphous state. These ternary dispersions exhibited superior solubility and dissolution performance in comparison to binary drug–mannitol systems and maintained structural integrity for up to six months under accelerated storage conditions (40 °C). Pesic et al. [30] developed NanoCrySP™, a spray-drying platform for converting nanosized drug particles into NSDs. This methodology has the potential to significantly enhance the formulation of active pharmaceutical ingredients (APIs) with polyol-based excipients compared to earlier methods. When combined with amino acids and contemporary spray-processing techniques, glucide and alditol systems can increase not just solubility but also drug stability and distribution within rationally planned pharmaceutical formulations.
Due to its advantageous properties, mannitol, a naturally occurring sugar alditol, is frequently used as a bulking agent in pharmaceutical formulations like lyophilized (freeze-dried) injectable systems. These advantages include low hygroscopicity and a high eutectic point in mannitol-water systems, which allows for primary drying in freeze-drying to occur at elevated temperatures [51]. In the area of freeze-dried solid dispersions (SDs), mannitol has been used to generate both partially and fully amorphous solid dispersions (ASDs), such as with lovastatin [13] and acetazolamide [14], respectively. In contrast to sugar-based carriers, mannitol-based ASDs have shown resistance to solution-mediated phase transitions and demonstrated physical stability for acetazolamide dispersions for up to six months under accelerated conditions (40±2 °C, 75±5%RH).
In addition to established workflows, co-grinding with mannitol has been tested as a possible option to limit the disadvantages of both fusion and solvent evaporation. This approach addresses the thermal damage to pharmaceuticals during fusion, where components have widely different melting temperatures, and the potential poisonous remnants of solvents from evaporation procedures. [45] Valsartan–mannitol SDs, developed through ball milling [26, 27], have demonstrated that both milling duration and speed are critical in achieving amorphization of the drug and carrier, typically requiring more than two hours. Mannitol’s inclusion significantly improved the dissolution rate of valsartan by enhancing wettability and reducing drug particle self-aggregation, compared to milling the drug alone. To solve the scalability limits of ball milling, Petry et al. [45] presented an air-jet mill-based continuous co-grinding approach. Their investigation on micronized griseofulvin-mannitol SDs revealed that the technique produced crystalline dispersions rather than amorphous forms. Nonetheless, both ball milling and air-jet milling improved drug release profiles by reducing crystallinity, increasing wettability, and ensuring uniform distribution of the active component within the carrier matrix. Remarkably, the mannitol-based SDs obtained from all these methods didn't exhibit solution-induced recrystallization, which is typical of sugar-based glucides. This remarkable behavior reinforces mannitol's reputation as a remarkable alditol capable of enhancing solubility and sustaining the release of drugs in pharmaceutical systems.
Sorbitol as a solubilizing agent
Sorbitol, a naturally occurring sugar alcohol and isomer of maltitol [34], is widely used in medicinal formulations due to its diverse functional characteristics. It has several functions in oral dosage forms, including filler, sweetener, quick disintegrant, stabilizer, and sugar substitute in syrups. Sorbitol also serves as a humectant in topical treatments and regulates isotonicity in a variety of injectable formulations [37]. Solid dispersions containing sorbitol generated by fusion or solvent evaporation, like mannitol, often yield crystalline suspensions. Notably, sorbitol has been shown to improve drug solubility and dissolution rates more effectively than mannitol [4] and several other hydrophilic carriers, including polyvinylpyrrolidone PVP K-30, polyethylene glycols PEG 6000, and hydroxypropyl methylcellulose (HPMC) [31, 17]. Similar to mannitol-based systems, the evaluated studies found no solution-mediated recrystallization. However, sorbitol's hygroscopic character, in contrast to mannitol [36] and other polyols [43], may present issues in terms of long-term physical stability of formulations.
Xylitol-based solid dispersions
Xylitol, a five-carbon polyhydric alcohol present in a range of fruits and vegetables, is one of the most commonly used sweetening agents in pharmaceutical applications [52]. It, like mannitol, was one of the first sugar alcohols to be studied as a carrier in solid dispersion (SD) systems with the goal of enhancing the solubility profiles of poorly soluble pharmaceuticals. Malkawi et al.'s [8] fundamental research indicated xylitol's applicability as a dispersion matrix, highlighting its beneficial properties such as a low melting point and strong thermal endurance. Although detailed insights into the molecular configurations of these xylitol-based SDs are limited, early findings demonstrated a remarkable enhancement-reporting up to a 200-fold increase in solubility for dispersions containing 5% butyl p-aminobenzoate. In subsequent studies, binary systems of famotidine and xylitol formed amorphous solid dispersions in which xylitol remained crystalline, while famotidine exhibited a broader endothermic profile in differential scanning calorimetry (DSC), indicative of reduced particle size. Unlike glucide-based dispersions, which frequently undergo recrystallization or multiphasic release, xylitol-containing formulations typically demonstrate monophasic release kinetics with no evidence.
Most xylitol SDs are produced using thermal techniques such as fusion or hot-melt extrusion (HME). These methods aid in solubilization and dissolution phenomena by enhancing the interactions of the aqueous medium with the drug particles and improving hydration of the drug surface [45, 50-53]. In comparative studies, xylitol often bested other polyols, sorbitol [36], and mannitol [17]. There is, however, a widespread concern regarding the xylitol formulations that centers on thermodynamic instability, which is generally the result of crystal formation during storage and can adversely affect the performance of the system over time. Xylitol has a glass transition temperature (Tg) of-19 °C to-24 °C [41], which means it will subsequently recrystallize when cooled to room temperatures. Nevertheless, solid dispersions in firm gelatin capsules showed sufficient shelf life of three months under moderate accelerant conditions (10). All in all, these studies substantiate the potential of alditol xylitol as a strong candidate for a solubilizing agent to improve the solubility and bioavailability of hydrophobic pharmaceutical compounds.
Erythritol-based solid dispersions
Erythritol, a naturally occurring four-carbon sugar alcohol, is frequently used as a bulk sweetener in food and pharmaceutical formulations due to its near-zero caloric content, excellent thermal resistance, and low water activity, making it essentially non-hygroscopic [10, 39, 40]. Despite its benefits, erythritol has limited aqueous solubility and tends to crystallize quickly [11]. Erythritol's low glass transition temperature (Tg) of around-45 °C [29] may explain the lack of studies employing it in solid dispersions (SDs) created using heat-cool cycles or lyophilization processes.
In one significant study, Jensen et al. [52] investigated the utilization of erythritol in ternary amorphous solid dispersions (ASDs) including griseofulvin and hypromellose phthalate and produced using hot-melt extrusion. The addition of erythritol increased the formulation's hydrophilic character while decreasing its viscoelasticity, resulting in improved drug solubilization and amorphous matrix stabilization, notably for the hypromellose phthalate component. Furthermore, erythritol has been shown to dissolve readily in organic solvents such as ethanol and methanol [9], implying its potential applicability in solid dispersions formulated using solvent evaporation techniques. This result opens a promising avenue for future research into its role as a natural alditol carrier in improving the dissolution performance and physical stability of poorly water-soluble active pharmaceutical ingredients.
Solid dispersions isomalt based
Isomalt consists of an equimolar blend of two disaccharide alcohols-α-D-glucopyranosyl-[1-6]-D-sorbitol (GPS) and α-D-glucopyranosyl-[1-6]-D-mannitol (GPM) [50]. It shares certain traits with erythritol, notably its extremely low moisture absorption. What sets isomalt apart from other polyols is its capacity to retain a glassy state after resolidification, a property linked to its relatively high glass transition temperature (Tg) of 61.5 °C, which is greater than any other known polyol [39]. This characteristic makes isomalt a promising carrier for solid dispersion (SD) systems, especially when applying the fusion method. However, the literature appears to lack research utilizing the fusion approach with isomalt.
Limited available studies on isomalt-based SDs have primarily used the spray-drying technique, typically employing ethanol as the solvent. The resulting SDs exhibit a glassy matrix, where the active pharmaceutical ingredient is converted into an amorphous form, and the particle size of isomalt is notably decreased. When comparing binary SDs (e. g., PVP and isomalt) to ternary systems (drug, PVP, and isomalt), the latter generally show enhanced performance in terms of solubility and dissolution rate. Studies assessing PVP versus isomalt as carrier materials have consistently shown higher formulation yield with isomalt [20].
Stability assessments of celecoxib [20] and indomethacin [2] demonstrated differing outcomes: celecoxib recrystallized after one month of storage, whereas indomethacin remained in its amorphous form. These findings may be attributed to the glass-forming tendencies of the drugs, where celecoxib exhibits moderate stability [6] and indomethacin is considered a strong glass former [13]. Moreover, the abundant hydroxyl groups in isomalt likely promote stronger molecular interactions with indomethacin, leading to improved physical stability. A notable challenge reported across these studies is the limited drug loading capacity, which typically ranges from 10% to 30%, often resulting in bulky final dosage forms.
Maltitol-based solid dispersions
Maltitol, a disaccharide-derived sugar alcohol, has physicochemical properties similar to those of isomalt. Chemically known as 4-O-β-D-glucopyranosyl-D-glucitol, it is made up of equal parts glucose and sorbitol [51]. This polyol has high aqueous solubility, similar to maltose [37], a moderate glass transition temperature (Tg) of around 49.5 °C, low moisture affinity, and several hydroxyl functionalities that facilitate drug-molecule interactions. Additionally, maltitol exhibits strong thermal endurance, reportedly decomposing at around 274 °C [41].
Despite its potential thermal and chemical properties, there appears to be a dearth of evidence on the use of maltitol in SD formulation using the fusion technique. Nonetheless, Imamura and colleagues [44, 46, 47] studied its use in the freeze-drying process to produce amorphous solid dispersions (ASDs). Maltitol-based systems did not significantly outperform other disaccharide carriers such as β-maltose, palatinose, and trehalose, but their contribution to ASD development remained significant. Its excellent physicochemical profile suggests that it could be further developed using various manufacturing procedures such as fusion processing and hot-melt extrusion (HME). The examined research highlights polyols' emerging importance as feasible alternatives to conventional sugars in SD development. However, selecting an acceptable polyol as a dispersion matrix is dependent on specific features, particularly glass transition behavior and crystallization kinetics, which in turn determine the best processing procedure. Although polyols such as xylitol, erythritol, isomalt, and maltitol have beneficial properties, their use is underreported, necessitating further examination in future studies.
Glucide and alditol co-systems
Drugs containing low molecular weight hydrophilic moieties, such as sugars and sugar alcohols, are often co-amorphous or co-crystalline complexes (also called "drug excipients") formed by drug formulations. The results of research indicate that these complexes exhibit greater dissolution and stability than purely crystalline or amorphous structures, thanks to intermolecular interactions like hydrogen bonds in the basal cells and π-stacking. Strong and specific molecular interactions between the drug and co-former in co-amorphous systems enhance stability by raising the glass transition temperature, preventing cryocrystallization. The availability of diverse glucides and alditols as co-formers gives a significant advantage over the normal counterions used in salt creation. Co-crystals can be designed to meet various properties such as phylogenetic and chemical stability, light resistance, permeability, compressibility, flow, solubility, or bioavailability while maintaining the action of the drug (although studies on these systems have limited data). However, polyol-based co-crystalline systems using sugars or polyochlorides are promising. Arafa and colleagues demonstrated that this was possible with cophosphate-type systems in Europe. Generally, co-crystals form single-phase solids characterized by a single melting peak located between those of the individual substances. The lack of single-crystal X-ray diffraction data, however, limits definitive proof of xylitol-based co-crystal formation. Utilizing the principles of supramolecular chemistry, polyols can form co-crystals due to their tendency to form extensive hydrogen bonding networks. It is hypothesized that heterosynthons between hydroxyl groups in polyols and drug functional groups, such as ethers, could facilitate co-crystal creation, but further experimental validation is necessary. These interactions reduce molecular mobility, elevate Tg, and suppress recrystallization, ensuring stability of amorphous dispersions.
Natural deep eutectic solvents
The growing demand for safer and more environmentally sustainable alternatives to conventional organic solvents has driven interest in green solvent systems across the pharmaceutical, food, and chemical industries. Among the promising candidates are ionic liquids (ILs), deep eutectic solvents (DESs), and natural deep eutectic solvents (NADESs), each exhibiting unique properties and challenges.
While ILs have been widely proposed as “green” solvents due to their negligible vapor pressure and tunable properties, concerns remain regarding their environmental persistence, toxicity, and complex synthesis. DESs, composed of hydrogen bond donors (HBDs) and acceptors (HBAs), have emerged as simpler, potentially less toxic alternatives, especially for enhancing drug solubility. However, the toxicity profiles of many synthetic DESs remain under-characterized, and their biocompatibility is not yet fully validated. These interactions reduce molecular mobility, elevate Tg, and suppress recrystallization, ensuring stability of amorphous dispersions.
NADESs, a subclass of DESs composed exclusively of naturally derived, biodegradable constituents-such as sugars, sugar alcohols, amino acids, and organic acids-have gained particular traction in pharmaceutical research. Owing to their low volatility, chemical tunability, thermal stability, and simple, solvent-free preparation, NADESs are often viewed as a more biocompatible and sustainable alternative to synthetic DESs. Nevertheless, broad claims of low toxicity require cautious interpretation. Narula A. et al. have demonstrated that NADESs’ safety profiles are highly dependent on their specific constituents and molar ratios [3]. Therefore, comprehensive in vitro and in vivo toxicological assessments are still needed before their widespread adoption in pharmaceutical formulations. Moreover, NADESs face technical formulation challenges, including high viscosity, which may limit processability and drug release rates, and difficulties in purification, particularly when residual components are difficult to remove or quantify. Despite these limitations, proof-of-concept studies have shown promising results. For example, Jeliński et al. [13, 32] demonstrated a substantial increase in the solubility of sulfanilamide (2-fold) and curcumin (up to 1000-fold) in NADES formulations based on choline chloride combined with sugars or polyols in various molar ratios. Similarly, Maugeri and De Maria [14] highlighted the pH neutrality of sugar-based NADESs, which is advantageous for pharmaceutical stability and compatibility with pH-sensitive APIs. Additionally, Malkawi R, et al. [8] successfully developed NADES-based transdermal patches, showcasing the solvents’ potential not only as solubilizers but also as functional excipients in advanced drug delivery systems. Such applications demonstrate that, with optimized formulation strategies, NADESs may serve as effective carriers for poorly water-soluble drugs, especially those classified under BCS Class II and IV.
In conclusion, while NADESs offer a sustainable and versatile platform for drug solubilization, their practical application in pharmaceuticals demands rigorous safety assessments, process optimization, and tailored formulation designs to address viscosity and scalability challenges.
CONCLUSION
Water-soluble excipients, particularly polyhydric alcohols (polyols), have emerged as superior carriers for solid dispersions due to their thermal stability, miscibility, and favorable molecular interactions compared to traditional sugars. Incorporating auxiliary excipients in ternary systems, such as leucine for stabilization and poloxamers for enhanced dissolution, further improves performance. However, regulatory concerns regarding elemental impurities in polyols require strict compliance with ICH Q3D. Natural polyols like isomalt, maltitol, xylitol, and erythritol show strong potential as biocompatible, taste-masking, and glass-forming carriers. Their integration into sustainable NADES-based platforms offers a promising pathway for next-generation oral drug delivery.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
Dalia A. Gaber; Conceptualization, Supervision, Project administration, Writing – review and editing. Mohammed A. Amin; Investigation, Data curation, Formal analysis, Writing – original draft, Visualization, Validation. Mostafa A. Mohamed; Software, Statistical analysis, Resources, Data interpretation, Writing – review and editing.
CONFLICT OF INTERESTS
Declared none
REFRENCES
Hebbink GA, Dickhoff BH. Application of lactose in the pharmaceutical industry. In: Watson RR, Preedy VR, Zibadi S, editors. Lactose. Amsterdam: Elsevier; 2019. p. 175-229. doi: 10.1016/B978-0-12-811720-0.00005-2.
Franca MT, Martins Marcos T, Costa PF, Bazzo GC, Nicolay Pereira RN, Gerola AP. Eutectic mixture and amorphous solid dispersion: two different supersaturating drug delivery system strategies to improve griseofulvin release using saccharin. Int J Pharm. 2022;615:121498. doi: 10.1016/j.ijpharm.2022.121498, PMID 35065207.
Narula A, Sabra R, Li N. Mechanisms and extent of enhanced passive permeation by colloidal drug particles. Mol Pharm. 2022;19(9):3085-99. doi: 10.1021/acs.molpharmaceut.2c00124, PMID 35998304.
Andrews GP, Qian K, Jacobs E, Jones DS, Tian Y. High drug loading nanosized amorphous solid dispersion (NASD) with enhanced in vitro solubility and permeability: benchmarking conventional ASD. Int J Pharm. 2023;632:122551. doi: 10.1016/j.ijpharm.2022.122551, PMID 36581107.
Lenhart A, Chey WD. A systematic review of the effects of polyols on gastrointestinal health and irritable bowel syndrome. Adv Nutr. 2017;8(4):587-96. doi: 10.3945/an.117.015560, PMID 28710145.
Grembecka M. Sugar alcohols. In: Reference module in food science. Amsterdam: Elsevier; 2018. p. 265-75.
De Stefani C, Lodovichi J, Albonetti L, Salvatici MC, Quintela JC, Bilia AR. Solubility and permeability enhancement of oleanolic acid by solid dispersion in poloxamers and β-cyclodextrin. Molecules. 2022;27(10):3042. doi: 10.3390/molecules27103042.
Malkawi R, Malkawi WI, Al Mahmoud Y, Tawalbeh J. Current trends on solid dispersions: past present and future. Adv Pharmacol Pharm Sci. 2022;2022:5916013. doi: 10.1155/2022/5916013, PMID 36317015.
Tambe S, Jain D, Meruva SK, Rongala G, Juluri A, Nihalani G. Recent advances in amorphous solid dispersions: preformulation formulation strategies technological advancements and characterization. Pharmaceutics. 2022;14(10):2203. doi: 10.3390/pharmaceutics14102203, PMID 36297638.
Zhang J, Guo M, Luo M, Cai T. Advances in the development of amorphous solid dispersions: the role of polymeric carriers. Asian J Pharm Sci. 2023;18(4):100834. doi: 10.1016/j.ajps.2023.100834, PMID 37635801.
Maher EM, Ali AM, Salem HF, Abdelrahman AA. In vitro / in vivo evaluation of an optimized fast dissolving oral film containing olanzapine co-amorphous dispersion with selected carboxylic acids. Drug Deliv. 2016;23(8):3088-100. doi: 10.3109/10717544.2016.1153746.
An JH, Lim C, Kiyonga AN, Chung IH, Lee IK, Mo K. Co-amorphous screening for the solubility enhancement of poorly water soluble mirabegron and investigation of their intermolecular interactions and dissolution behaviors. Pharmaceutics. 2018;10(3):149. doi: 10.3390/pharmaceutics10030149, PMID 30189645.
Zhang M, Suo Z, Peng X, Gan N, Zhao L, Tang P. Microcrystalline cellulose as an effective crystal growth inhibitor for the ternary ibrutinib formulation. Carbohydr Polym. 2020;229:115476. doi: 10.1016/j.carbpol.2019.115476, PMID 31826488.
Maincent J, Williams RO 3rd. Sustained release amorphous solid dispersions. Drug Deliv Transl Res. 2018;8(6):1714-25. doi: 10.1007/s13346-018-0494-8, PMID 29498004.
Knopp MM, Wendelboe J, Holm R, Rades T. Effect of amorphous phase separation and crystallization on the in vitro and in vivo performance of an amorphous solid dispersion. Eur J Pharm Biopharm. 2018;130:290-5. doi: 10.1016/j.ejpb.2018.07.005, PMID 30064702.
Motallae S, Taheri A, Homayouni A. Preparation and characterization of solid dispersions of celecoxib obtained by spray-drying ethanolic suspensions containing PVP-K30 or isomalt. J Drug Deliv Sci Technol. 2018;46:188-96. doi: 10.1016/j.jddst.2018.05.020.
Apiwongngam J, Limwikrant W, Jintapattanakit A, Jaturanpinyo M. Enhanced supersaturation of chlortetracycline hydrochloride by amorphous solid dispersion. J Drug Deliv Sci Technol. 2018;47:417-26. doi: 10.1016/j.jddst.2018.08.007.
Mizoguchi R, Waraya H, Hirakura Y. Application of co-amorphous technology for improving the physicochemical properties of amorphous formulations. Mol Pharm. 2019;16(5):2142-52. doi: 10.1021/acs.molpharmaceut.9b00105, PMID 30946778.
Meng Lund H, Kasten G, Jensen KT, Poso A, Pantsar T, Rades T. The use of molecular descriptors in the development of co-amorphous formulations. Eur J Pharm Sci. 2018;119:31-8. doi: 10.1016/j.ejps.2018.04.014, PMID 29649569.
Pajula K, Hyyrylainen J, Koistinen A, Leskinen JT, Korhonen O. Detection of amorphous amorphous phase separation in small molecular co-amorphous mixtures with SEM-EDS. Eur J Pharm Biopharm. 2020;150:43-9. doi: 10.1016/j.ejpb.2020.03.002, PMID 32151730.
Wostry M, Plappert H, Grohganz H. Preparation of co-amorphous systems by freeze-drying. Pharmaceutics. 2020;12(10):941. doi: 10.3390/pharmaceutics12100941, PMID 33008124.
Bhalani DV, Nutan B, Kumar A, Singh Chandel AK. Bioavailability enhancement techniques for poorly aqueous soluble drugs and therapeutics. Biomedicines. 2022;10(9):2055. doi: 10.3390/biomedicines10092055, PMID 36140156.
Aimurofiq A, Putro DS, Ramadhani DA, Putra GM, Espirito Santo LDC. A review on solubility enhancement methods for poorly water-soluble drugs. J Rep Pharm Sci. 2021;10(3):137-47. doi: 10.4103/jrptps.JRPTPS_134_19.
Rahman Z, Wengel J, Rades T, Lobmann K. Molecular structure and impact of amorphization strategies on intrinsic dissolution of spray dried indomethacin. Int J Pharm. 2019;569:118636. doi: 10.1016/j.ijpharm.2019.118636.
Tekade AR, Yadav JN. A review on solid dispersion and carriers used therein for solubility enhancement of poorly water soluble drugs. Adv Pharm Bull. 2020;10(3):359-69. doi: 10.34172/apb.2020.044, PMID 32665894.
Vanda H, Verpoorte R, Klinkhamer PG, Choi YH. Natural deep eutectic solvents: from their discovery to their applications. In: Ramon DJ, Guillena G, editors. Deep eutectic solvents: synthesis properties and applications. Weinheim: Wiley-VCH Press; 2019. p. 61-81. doi: 10.1002/9783527818488.ch4.
Jelinski T, Przybyłek M, Cysewski P. Solubility advantage of sulfanilamide and sulfacetamide in natural deep eutectic systems: experimental and theoretical investigations. Drug Dev Ind Pharm. 2019;45(7):1120-9. doi: 10.1080/03639045.2019.1597104, PMID 30883240.
Jelinski T, Przybylek M, Cysewski P. Natural deep eutectic solvents as agents for improving solubility stability and delivery of curcumin. Pharm Res. 2019;36(8):116. doi: 10.1007/s11095-019-2643-2, PMID 31161340.
Dai Y, Van Spronsen J, Witkamp GJ, Verpoorte R, Choi YH. Natural deep eutectic solvents as new potential media for green technology. Anal Chim Acta. 2013;766:61-8. doi: 10.1016/j.aca.2012.12.019, PMID 23427801.
Liu Y, Friesen JB, McAlpine JB, Lankin DC, Chen SN, Pauli GF. Natural deep eutectic solvents: properties applications and perspectives. J Nat Prod. 2018;81(3):679-90. doi: 10.1021/acs.jnatprod.7b00945, PMID 29513526.
Weerapol Y, Tubtimsri S, Jansakul C, Sriamornsak P. Improved dissolution of Kaempferia parviflora extract for oral administration by preparing solid dispersion via solvent evaporation. Asian J Pharm Sci. 2017;12(2):124-33. doi: 10.1016/j.ajps.2016.09.005, PMID 32104321.
Kauppinen A, Broekhuis J, Grasmeijer N, Tonnis W, Ketolainen J, Frijlink HW. Efficient production of solid dispersions by spray drying solutions of high solid content using a 3-fluid nozzle. Eur J Pharm Biopharm. 2018;123:50-8. doi: 10.1016/j.ejpb.2017.11.009, PMID 29162509.
Wong WS, Lee CS, Er HM, Lim WH, Wong SF. Biocompatible palm stearin-based polyesteramide as polymer carrier for solid dispersion. J Appl Polym Sci. 2018;135(8):45892. doi: 10.1002/app.45892.
Gaikwad D, Shewale R, Patil V, Mali D, Gaikwad U, Jadhav N. Enhancement in in vitro anti-angiogenesis activity and cytotoxicity in lung cancer cell by pectin-PVP based curcumin particulates. Int J Biol Macromol. 2017;104(A):656-64. doi: 10.1016/j.ijbiomac.2017.05.170, PMID 28602990.
Lenz E, Lobmann K, Rades T, Knop K, Kleinebudde P. Hot melt extrusion and spray drying of co-amorphous indomethacin arginine with polymers. J Pharm Sci. 2017;106(1):302-12. doi: 10.1016/j.xphs.2016.09.027, PMID 27817830.
Petry I, Lobmann K, Grohganz H, Rades T, Leopold CS. Solid state properties and drug release behavior of co-amorphous indomethacin arginine tablets coated with Kollicoat® protect. Eur J Pharm Biopharm. 2017;119:150-60. doi: 10.1016/j.ejpb.2017.06.007, PMID 28602869.
Petry I, Lobmann K, Grohganz H, Rades T, Leopold CS. Undesired co-amorphisation of indomethacin and arginine during combined storage at high humidity conditions. Int J Pharm. 2018;544(1):172-80. doi: 10.1016/j.ijpharm.2018.04.026, PMID 29669257.
Petry I, Lobmann K, Grohganz H, Rades T, Leopold CS. In situ co-amorphisation in coated tablets the combination of carvedilol with aspartic acid during immersion in an acidic medium. International Journal of Pharmaceutics. 2019;558:357-66. doi: 10.1016/j.ijpharm.2018.12.091.
Lim AW, Lobmann K, Grohganz H, Rades T, Chieng N. Investigation of physical properties and stability of indomethacin cimetidine and naproxen cimetidine co-amorphous systems prepared by quench cooling coprecipitation and ball milling. J Pharm Pharmacol. 2016;68(1):36-45. doi: 10.1111/jphp.12494, PMID 26663364.
Russo MG, Sancho MI, Silva LM, Baldoni HA, Venancio T, Ellena J. Looking for the interactions between omeprazole and amoxicillin in a disordered phase. An experimental and theoretical study. Spectrochim Acta A Mol Biomol Spectrosc. 2016;156:70-7. doi: 10.1016/j.saa.2015.11.021, PMID 26654963.
Mishra J, Lobmann K, Grohganz H, Rades T. Influence of preparation technique on co-amorphization of carvedilol with acidic amino acids. Int J Pharm. 2018;552(1-2):407-13. doi: 10.1016/j.ijpharm.2018.09.070, PMID 30278256.
Liu J, Grohganz H, Rades T. Influence of polymer addition on the amorphization dissolution and physical stability of co-amorphous systems. Int J Pharm. 2020;588:119768. doi: 10.1016/j.ijpharm.2020.119768, PMID 32798592.
Mishra J, Rades T, Lobmann K, Grohganz H. Influence of solvent composition on the performance of spray-dried co-amorphous formulations. Pharmaceutics. 2018;10(2):47. doi: 10.3390/pharmaceutics10020047, PMID 29649124.
Jensen KT, Blaabjerg LI, Lenz E, Bohr A, Grohganz H, Kleinebudde P. Preparation and characterization of spray-dried co-amorphous drug amino acid salts. J Pharm Pharmacol. 2016;68(5):615-24. doi: 10.1111/jphp.12458, PMID 26245703.
Kasten G, Duarte I, Paisana M, Lobmann K, Rades T, Grohganz H. Process optimization and upscaling of spray-dried drug amino acid co-amorphous formulations. Pharmaceutics. 2019;11(1):24. doi: 10.3390/pharmaceutics11010024, PMID 30634423.
Daravath B, Naveen C, Vemula SK, Tadikonda RR. Solubility and dissolution enhancement of flurbiprofen by solid dispersion using hydrophilic carriers. Braz J Pharm Sci. 2017;53(4):e00010. doi: 10.1590/S2175-97902017000400010.
Hattali WS, Samuel BA, Philip AK. Enhancing fluconazole solubility and bioavailability through solid dispersion techniques: evaluation of polyethylene glycol 6000 and sodium carboxymethylcellulose systems. Int J Pharm Pharm Sci. 2024;16(12):51-9. doi: 10.22159/ijpps.2024v16i12.52739.
Puppala RK, A VL. Optimization and solubilization study of nanoemulsion budesonide and constructing pseudoternary phase diagram. Asian J Pharm Clin Res. 2019;12(1):551-3. doi: 10.22159/ajpcr.2019.v12i1.28686.
Wood CC, Patel KG, Weber VL, Osakwe AR, Manovacia Moreno NPM, Broich ML. Development of impact resistant immediate release amorphous solid dispersion via hot-melt extrusion and injection molding. Int J Pharm. 2025;680:125746. doi: 10.1016/j.ijpharm.2025.125746, PMID 40449639.
Wu J, Den Mooter GV. Statistical analysis of long-term physical stability testing of amorphous solid dispersions. Int J Pharm. 2025;681:125844. doi: 10.1016/j.ijpharm.2025.125844, PMID 40517971.
Mishra J, Rades T, Lobmann K, Grohganz H. Influence of solvent composition on the performance of spray-dried co-amorphous formulations. Pharmaceutics. 2018;10(2):47. doi: 10.3390/pharmaceutics10020047, PMID 29649124.
Hattali WS, Samuel BA, Philip AK. Enhancing fluconazole solubility and bioavailability through solid dispersion techniques: evaluation of polyethylene glycol 6000 and sodium carboxymethylcellulose systems. Int J Pharm Pharm Sci. 2024;16(12):51-9. doi: 10.22159/ijpps.2024v16i12.52739.
Kasten G, Duarte I, Paisana M, Lobmann K, Rades T, Grohganz H. Process optimization and upscaling of spray-dried drug amino acid co-amorphous formulations. Pharmaceutics. 2019;11(1):24. doi: 10.3390/pharmaceutics11010024, PMID 30634423.