
Int J App Pharm, Vol 17, Issue 6, 2025, 302-311Original Article
FORMULATION AND EVALUATION OF EFFERVESCENT GRANULES CONTAINING ANTIRETROVIRAL AGENT SOLID DISPERSIONS FOR CHILDREN AND ELDERS: SOLUBILITY ENHANCEMENT, DISSOLUTION RATE AND INTEGRATION INHIBITION EFFECT
SUNITHA SAMPATHI1, LAKSHMI DEVI GOTTEMUKKULA2*, B. AISHWARYA2, M. JYOTHIRADITYA2, B. MANIKANTH2
1Department of Pharmacy, Vishwakarma University, Pune, Maharashtra, India. 2Joginpally B. R. Pharmacy College, Moinabad, R. R. District, Hyderabad, Telangana-500075, India
*Corresponding author: Lakshmi Devi Gottemukkula; *Email: s.sunitha@vupune.ac.in
Received: 23 Jun 2025, Revised and Accepted: 09 Oct 2025
ABSTRACT
Objective: Dolutegravir inhibits viral replication by blocking the integrase strands. To treat human immunodeficiency virus (HIV), it is one of the medications used in children and the elderly. It is difficult to get a high oral bioavailability of dolutegravir because of these population issues with swallowing and ineffective absorption, as well as the drug's intrinsic poor solubility and instability in stomach acid. To address these problems, we created and described effervescent granules containing a solid dolutegravir dispersion.
Methods: The melt granulation process was used with various categories and quantities of polymers to develop solid dispersion-based effervescent granules. The solid dispersions were prepared using four different methods: Physical Mixing, Co-grinding, Kneading and fusion methods. Drug-to-carrier ratios of 1:1, 1:2, 1:3, and 1:4 were investigated. To develop solid dispersions, three generations of polymers are selected [crystalline carrier (mannitol), Amorphous carriers [polyethene glycol (PEG 4000), polyvinyl pyrrolidone (PVP K90), hydroxypropyl methylcellulose (HPMC E5LV), sulfobutylether-Captisol (captisol)], poloxamer 188 and poloxamer 407].
Results: The DTG and the chosen polymer are compatible, according to FTIR studies; DSC and XRD results reveal crystalline changes of DTG; surface morphology reveals that pure DTG has a crystalline prismatic shape. Second, a total of 12 formulations of the effervescent granules containing the solid dispersion were developed using various excipients such as sweeteners, gas generators, pH modulators, and glidants/lubricants. The results show that all 12 formulations had a good release profile within 5 min. F3 shows the highest drug content (99.5±0.11), the highest percentage of drug release 99.5% within 5 min and the effervescent cessation time 19.5±2.12 sec. The good release profile of F3 may be attributed to the combination of cetyl alcohol and sodium starch glycolate.
Conclusion: Additional in vivo and clinical research may be conducted on the final effervescent granule product to develop it into a dolutegravir delivery system with high bioavailability for older people and children.
Keywords: Captisol®, Effervescent granules, Fusion method, Solid dispersions, Solubility enhancement
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2025v17i6.55697 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Human Immunodeficiency Virus (HIV), originating from simian immunodeficiency virus (SIV), infects human immune system cells, leading to compromised immune systems and AIDS. Dolutegravir (DTG), a second-generation HIV integrase inhibitor, is effective against resistance and is used in pre-exposure prophylaxis. DTG is more effective than other integrase strand transfer inhibitors (ISTIs), which may gain resistance. It is a prevention tool for those at substantial risk of contracting HIV [1].
Paediatric pharmacology faces challenges in optimising oral drug delivery due to children's discomfort with swallowing solid dosage forms. Studies show 54% of children aged 6-11 struggle to swallow tablets easily [2]. However, recent improvements in pediatric trials have been made due to increased pediatric expertise and US legislation mandating children's studies. The European Union's 2007 pediatric medicinal regulations have further stimulated pediatric drug development, particularly in Europe [3, 4].
Due to the WHO’s efforts, some success has been achieved for “tropical” diseases such as malaria and tuberculosis, where pediatric patient-friendly suspension and dispersible dosage forms are becoming available [5, 6]. However, the availability of such dosages is still waited for antiretroviral drugs, as HIV/AIDS are the major challenges in these countries [7, 8]. At present, fixed-dose combinations (FDCs) and once daily solid dosage forms are helping adolescents and older children. Still, suitable dosage forms are lacking for younger children and infants, which results in poor adherence, viral resistance, and decreased survival of HIV-positive children [9].
Dolutegravir, a drug with poor solubility and bitter taste, is a potential solution for children and the elderly due to its difficulty in swallowing and inefficient absorption [10]. Effervescent granules with solid dispersion, combined with hydrophilic polymers, can improve solubility and bioavailability [11]. Solid dispersion systems, such as physical mixing, co-grinding, kneading, and fusion, offer advantages such as less organic solvents, drug decomposition avoidance, and homogenization of drugs and polymers [12, 13].
Several research papers have been published on improving Dolutegravir's physico-chemical properties using solid dispersion [14]. For instance, Sundeep Mupparaju (2021) prepared the dolutegravir solid dispersion with a poloxamer-188 carrier using the fusion method, with a 5.11-times increase in the dolutegravir solubility [15, 16]. Monika Bhairam (2022) successfully prepared Dolutegravir solid dispersion with Soluplus and proved increased solubility and bioavailability compared to pure API [17, 18]. Additionally, S. Chaudhary (2021) formulated solid dispersions using poloxamer 407 and performed in vivo evaluation; based on the findings, solid dispersions offer an increased release rate and improved bioavailability [19]. S. Martha (2021) formulated solid dispersion-based fast dissolving tablets to enhance solubility and dissolution rate using PEG 6000 and poloxamer 407 [20].
Nevertheless, to our knowledge, no studies have considered the pediatric and elderly dosage forms of dolutegravir solid dispersion and evaluated the system's ability to protect the drug from the gastric acidic condition. To that end, this study developed and characterized the effervescent granules containing dolutegravir solid dispersion to enhance the dolutegravir solubility [14, 2] decrease the drug bitterness [21], and counteract the effects of acidic pH in the stomach environment [22]. Firstly, the DTG solubility at different pH levels was evaluated. Then, the solid dispersions, at different polymers and concentrations, were prepared by the physical mixing method, co-grinding method, kneading method and fusion method [21]. The optimised formulations are fully characterised. The optimal formula was then fabricated to be the effervescent granules, taking into account the types and amounts of numerous excipients. Finally, the product properties were determined based on the Vietnamese Pharmacopoeia.
MATERIALS AND METHODS
Materials
Dolutegravir (DTG) was received as a gift sample from Styrax Pharma Pvt. Ltd, J. N. Pharma City, Anakapalli, India. Poloxamer 188, Poloxamer 407 were obtained as gift samples from BASF, INDIA Ltd. Hydroxy propyl methyl cellulose E5LV (HPMCE5LV), Captisol® (Sulfobutylether Captisol®sodium salt), Polyvinyl Pyrrolidone K90 (PVPK90), Polyethylene Glycol (PEG 4000), cetyl alcohol, sodium starch glycolate, citric acid, tartaric acid, saccharin sodium carbonate, sodium hydro carbonate, calcium carbonate, sulfuric acid, hydrochloric acid, potassium dihydrophosphate, and phosphoric acid were purchased from Asian chemicals, Hyderabad. All other reagents, chemicals, and solvents were of pharmaceutical grade or higher.
Methodology saturation solubility study
The drug solubility at different pH levels of 1.2, 6.0, 6.8, and 7.0, simulating different parts of the gastrointestinal tract (i. e., the stomach, the duodenum, the small intestine, and the large intestine), was evaluated using the shaking method during 120 min. For this, an excess amount of dolutegravir was added to 10 ml of the respective media to get the saturated solution, and the solution was shaken at 100 rpm at a temperature of 37±0.5 °C [23]. The bottle was placed in a trembling bath. Constant temperature was held at about 37±0.5 °C throughout the process, and shaking was performed at 50 rpm for 48 h. The samples are then filtered using a 0.45 mm filter paper. The filtrate was then analysed using a 260 nm UV-Vis spectrophotometer (UV 1700, Shimadzu, Japan) [24]. The dolutegravir solubility was then calculated based on the standard curve (y = 0.0067x+0.0554, R =0.9999) and reported.
Phase solubility study
Phase solubility studies with various carriers, including PEG 4000, PVPK90, Poloxamer 188, Poloxamer 407, Captisols, and HPMC E5 lV, were used to determine the best carrier for Dolutegravir. Distilled water was used to generate solutions from different concentrations, as shown in fig. 1 [25]. Excess amounts of DTG were mixed into glass vials with all the aqueous polymer solution before shaking for 72 h on a biological shaker (Orbital shaker SI 300, Jeio Tech, Korea) at room temperature. A 0.45 mm membrane filter was used to filter the contents after centrifuging them at 4000 rpm for 15 min. Spectrophotometric analysis (UV-visible spectrophotometer-UV 1700, Shimadzu, Japan) was used to determine the solubility at 260 nm [12, 26].
Preparation methods of dolutegravir solid dispersions
Based on the phase solubility studies, the optimised polymers were used independently to fabricate dolutegravir solid dispersion. Accordingly, four ratios of drug: polymer (w/w) were applied for each polymer [22]. These ratios were selected based on the previous reports and our preliminary results. Additionally, four preparation methods were employed: Physical mixing, co-grinding, kneading, and fusion. Thus, a total of 16 formulas were prepared, which are shown in table 1. For the physical mixing technique, accurately weigh the drug and polymer, thoroughly mix them with a spatula for 30 min, and pass through a No. 100 sieve. Table 1 details the drug and polymer ratios to prepare solid dispersions [27]. For Co-grinding technique, the carrier and drug were combined in a mortar with weighed amounts, co-ground for 20 min, and then sifted through a sieve no 100 [23] and in Kneading technique Weighing and placing the necessary quantities of drug and carrier in a mortar, the mixture was kneaded for 20 min with 70% v/v methanol (1.5 times by w/v). The resulting mass was ground up, dried at 40 degrees Celsius, and sifted through a sieve No. 100 [28]. In the fusion method, Optimised polymer is placed in a china dish and heated to 65 °C until it melts, then dolutegravir is added. After vigorous stirring at normal temperature, the mixture solidifies. The solid mass was crushed, pulverised and extruded through a 0.2-mm sieve to obtain the solid dispersion [16]. The granules obtained were stored in a desiccator for further studies.
Table 1: Formulation of dolutegravir solid dispersions
| Code | Composition | Ratio | Method |
| F1-F4 | Drug+Captisol | 1:1,1:2,1:3 and 1:4 | Physical mixing method |
| F5-F8 | Drug+Captisol | 1:1,1:2,1:3 and 1:4 | Co-grinding method |
| F9-F12 | Drug+Captisol | 1:1,1:2,1:3 and 1:4 | Kneading method |
| F13-F16 | Drug+Captisol | 1:1,1:2,1:3 and 1:4 | Fusion method |
Physicochemical evaluations of dolutegravir solid dispersions
Morphology, DSC, XRD and IR analyses
The dolutegravir solid dispersions were morphologically observed using the SEM. For this, 2–3 mg of the dispersion powders was sprayed onto a metal disc covered with conductive double-sided tape to create a slight layer of powder, followed by gold coating. SEM images of the surfaces of the particles were captured and observed in the vacuum chamber [29]. For the DSC measurements, 50–100 mg of dolutegravir solid dispersion powders were weighed and scattered on a DSC aluminium pan to form a flat, solid layer 2 mm in height. Then, the lid, pre-punched with a tiny hole, was subjected to the pan. A reference pan was prepared similarly, without the solid dispersion. Finally, the two aluminium pans were put in the heating chamber and analysed in a temperature range of 30–250 °C at an increment of 5 °C/min [30]. For the IR analysis, 1–2 mg of the samples (pure Dolutegravir powder, the pure polymers, and the Dolutegravir solid dispersion) were ground and mixed with 300–400 mg of KBr. The homogenised mixture was then compressed into 13-mm-diameter tablets with an 800-MPa pressing force. The IR analyses were performed with the received tablets, with a wavenumber range of 4000-400 cm-1 [31]. X-Ray Powder Diffraction (XRPD) patterns were obtained using a high-resolution X-ray diffractometer (Malvern Panalytical Empyrean 3), with a scanning angle of 0-40° of 2θ at 40 kV with Cu Kα radiation [32].
Percentage yield
The percentage yield of the prepared solid dispersions was calculated by using the following formula [11].
% Yield = 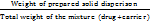 × 100
× 100
In vitro dissolution studies
In vitro dissolution studies used a USP Type-II (model DS 8000, Lab India, Mumbai, India) paddle-type dissolution test apparatus. A 50 mg equivalent weight of dolutegravir solid dispersion was enclosed in a capsule and placed in 900 ml of pH 6.8 phosphate buffer. Aliquots were withdrawn periodically and replaced with fresh buffer to maintain sink conditions. The samples were filtered using a 0.45 μm filter paper and analysed using a UV-visible spectrophotometer (UV 1700, Shimadzu, Japan) at 260 nm [12, 33].
Stability studies
Stability studies were conducted on the optimised Dolutegravir solid dispersion formulation F16. These formulations were packed in HDPE bottles with child-resistant caps (CRC) and induction sealed. The bottles were then subjected to accelerated stability conditions at 40 °C and 75% relative humidity [24]. Samples were evaluated for % assay and drug periodically at 3 mo and 6 mo.
Preparation of effervescent granules containing dolutegravir solid dispersion
Formulation procedure
Dolutegravir solid dispersions based on effervescent granules were prepared by the melt granulation technique, also known as thermoplastic granulation [34]. The granulation was achieved by adding a meltable binder in a solid state at room temperature, but preferably melts in the temperature range of 50 °C-80 °C. Further, no liquid binder or water was required as the binder in the molten state acts as a granulating liquid, and dried granules can be obtained easily by cooling at room temperature. Meltable binder such as Cetyl alcohol of concentrations 5%, 7.5% and 10% was used in the formulation [35]. Active pharmaceutical ingredient, i. e., equivalent weight of Dolutegravir solid dispersion and all other excipients, was weighed, and geometrical mixing was done. All the above ingredients were heated at about 50-80 °C in a heating mantle until a molten state was formed. Then it was cooled down at room temperature and passed through a sieve number 20 to obtain granules, and finally the granules were dried at a temperature not more than 60 °C in a hot air oven [36]. Below (table 2) shows the formulations of effervescent granules and solid dispersions containing dolutegravir.
Table 2: Formulation table of dolutegravir effervescent granules
| Formul-ation | E. SD (mg) | Cetyl alcohol (mg) |
Ssg (mg) | Citric acid (mg) | Tartaric acid (mg) | NaHCO3 (mg) | Saccharin | Sodium carbonate |
| F1 | 10 | 0. 25 | 3 | 26.25 | 0.23 | 24.00 | 0.15 | 11.38 |
| F2 | 10 | 0.5 | 2.75 | 26.25 | 0.23 | 24.00 | 0.15 | 0.88 |
| F3 | 10 | 0.75 | 2.5 | 26.25 | 0.23 | 24.00 | 0.15 | 2.96 |
| F4 | 10 | 1.0 | 2.25 | 26.25 | 0.23 | 24.00 | 0.15 | 6.88 |
| F5 | 10 | 1.25 | 2 | 26.25 | 0.23 | 24.00 | 0.15 | 6.13 |
| F6 | 10 | 1.5 | 1.75 | 26.25 | 0.23 | 24.00 | 0.15 | 6.13 |
| F7 | 10 | 1.75 | 1.5 | 26.25 | 0.23 | 24.00 | 0.15 | 6.13 |
| F8 | 10 | 2.0 | 1.25 | 26.25 | 0.23 | 24.00 | 0.15 | 9.31 |
| F9 | 10 | 2.25 | 1 | 26.25 | 0.23 | 24.00 | 0.15 | 6.13 |
| F10 | 10 | 2.5 | 0.75 | 26.25 | 0.23 | 24.00 | 0.15 | 6.13 |
| F11 | 10 | 2.75 | 0.5 | 26.25 | 0.23 | 24.00 | 0.15 | 1.88 |
| F12 | 10 | 3.0 | 0.25 | 26.25 | 0.23 | 24.00 | 0.15 | 5.38 |
Evaluation of dolutegravir effervescent granules
Angle of repose
Angle of repose is the maximum angle between the surface of the pile of powder and the horizontal plane. The lower the angle of repose, the better the flow properties. The angle of response is designated by ‘θ’ and given by the following equation [30].
Tan θ = h/r
Where h = height of the pile
r = radius of the base of the pile
Bulk Density, tapped density, compressibility index (Carr’s index) and Hausner’s ratio
10 g of granules was taken in a graduated measuring cylinder, and the initial volume was measured. A measuring cylinder containing granules was tapped until no further volume changes occurred [37]. Calculation was done using the following equations:
Bulk density =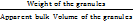
Tapped density =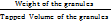
Compressibility Index (CI) = 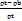 x 100
x 100
Where, ρt = Tapped density
ρb = Bulk density
Hausner’s ratio = 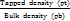
Effervescence cessation time
In vitro effervescence cessation time was measured by pouring one dose of granules in a 250 ml beaker containing 100 ml of distilled water. Granules from each batch were selected, and the in vitro effervescence time was measured [17].
Drug content
A sample equivalent to 50 mg of DTG was precisely weighed and transferred to a 100 ml volumetric flask. To ensure complete solubilization of DTG, 1 ml of methanol was added to the flask. This step is crucial as DTG has limited aqueous solubility, and methanol acts as a co-solvent to facilitate complete dissolution [22]. The volume was then made up to 100 ml using pH 6.8 phosphate buffer, which mimics the intestinal pH where drug absorption primarily occurs. The resulting solution was thoroughly mixed to ensure homogeneity. The concentration of DTG in this solution was then determined using a UV-visible spectrophotometer at a wavelength of 260 nm [1], which corresponds to the maximum absorption (λmax) of DTG. The absorbance values were compared against a pre-established calibration curve to calculate the drug content. This method accurately determines drug loading efficiency in the solid dispersion formulations and ensures batch-to-batch consistency [2].
In vitro dissolution study
The dissolution study of dolutegravir solid dispersion-based granules was done using the USP type II dissolution test apparatus. The dissolution test was performed using a dissolution medium made of phosphate buffer with pH 6.8 and at 37±0.5ᵒC and 50 rpm. A sample of 5 ml will be drawn every 1-minute interval and then replenished with 5 ml to maintain the constant volume. After that, the sample was filtered through a Whitman filter paper, and then the absorbance of the sample was measured at 260 nm. Then, the amount of drug released was calculated from a previously prepared calibration curve of dolutegravir [15].
RESULTS AND DISCUSSION
Evaluation of saturation solubility of dolutegravir in various pH buffers
The solubility of Dolutegravir was determined sequentially in the solutions with pH 1.2, 6.0, 6.8, and 7.0. dolutegravir (DTG) is a weak acid. It has a pKa of 8.2, which means it is only partially ionised in solution [15]. Tricyclic Metal-Chelating Core: This core is crucial for dolutegravir's binding to the HIV-1 integrase enzyme's active site. It contains oxygen atoms that chelate (bind tightly) to divalent metal ions, playing a role in stabilising the complex and facilitating the inhibition of viral DNA integration. The dolutegravir becomes more soluble as pH rises because of a tricyclic metal chelating moiety. Conclusively, at both low and high pH, the Dolutegravir oral bioavailability is considerably inadequate. To overcome this issue, Dolutegravir solid dispersions were utilised.
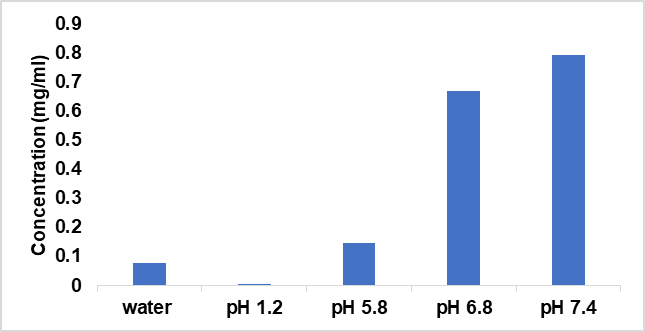
Fig. 1: Saturation solubility of dolutegravir in various pH buffers
Phase solubility studies
The phase solubility study was conducted to determine the best carrier for enhancing the solubility of dolutegravir (DTG). Various carriers were evaluated, including PEG 4000, PVPK90, Poloxamer 188, Poloxamer 407, Captisols, and HPMC E5 lV. The results showed improved solubility (fig. 1) of DTG with certain carriers, particularly captisol, and hence this was selected for further studies [35, 40]. Based on phase solubility studies, Captisol was selected as a suitable carrier for the formulation of solid dispersion. The aqueous solubility of Dolutegravir was found to be 15.28 μg/ml. Captisolin increased the solubility of DLG to 32670μg/ml in 10% aqueous solution. This indicated an increase of 275 times; Captisol were used for further studies.
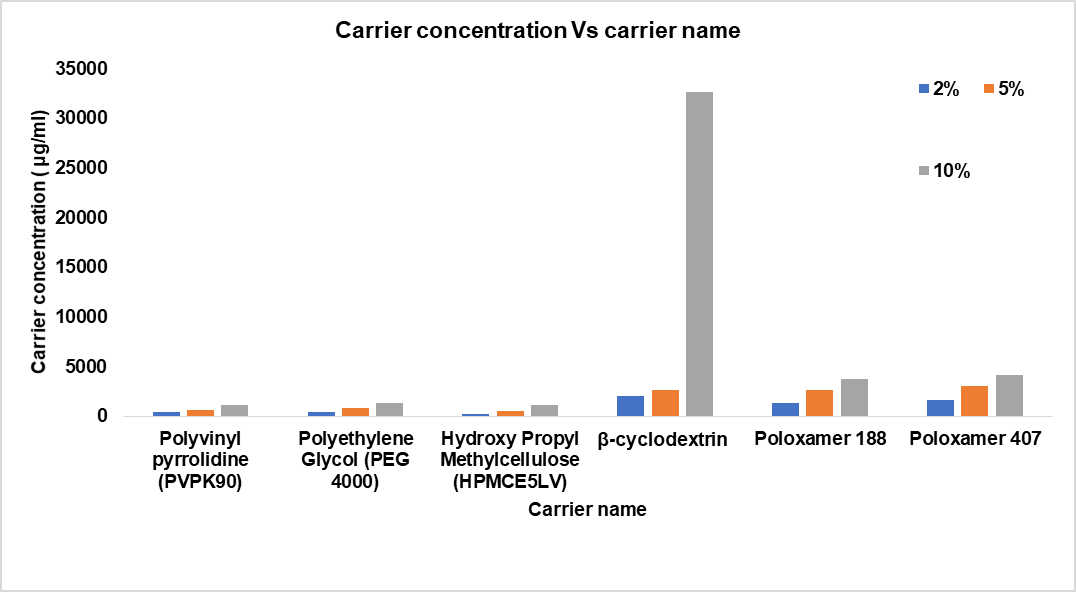
Fig. 2: Phase solubility studies in various carriers with different percentages
Morphology, XRD, DSC, and IR analyses
To characterise the Dolutegravir solid dispersion and to elucidate the interactions between Dolutegravir and β-CD, IR, SEM, XRD and DSC analytical techniques were employed.
Scanning electron microscopy (SEM)
SEM micrographs of pure Dolutegravir (fig. 2a) revealed irregularly shaped, crystalline particles with sharp edges and a rough surface texture. The particle size ranged from approximately 5-20 μm. In contrast, formulation F16 (fig. 2b) showed smaller, more rounded particles with less defined edges, ranging from 2-10 μm in size. The surface appeared smoother than the pure drug, indicating that some particles are coated around the dolutegravir particles. The particles appeared as thin, flaky structures with a highly porous surface. Individual particle boundaries were less distinct, forming a network-like structure. This unique morphology, characteristic of fused products, presented a significantly larger surface area than the other formulations, which could explain its superior dissolution performance. In solid dispersion formulations (F16), the distinct crystalline features of pure Dolutegravir were no longer visible, supporting the XRPD results indicating amorphisation [13, 32].

Fig. 2a: Scanning electron microscopy (SEM) image of the pure dolutegravir

Fig. 2b: The SEM image of dolutegravir solid dispersion with Captisol®at a ratio of 1:4 w/w
X-Ray powder diffraction (XRPD)
The XRPD pattern of pure Dolutegravir exhibited sharp, intense peaks at approximately 2θ values of 9.5°, 14.8°, 18.2°, 21.6°, 25.3° and 28.1°, characteristic of its crystalline nature. These distinct peaks indicate a highly ordered crystal structure of the pure drug. In contrast, the XRPD pattern of formulation F16 revealed a complete absence of sharp diffraction peaks. Instead, a broad, diffuse halo was observed in the 2θ range of 20-25°, characteristic of an amorphous material. This pattern strongly suggests the successful conversion of crystalline Dolutegravir to an amorphous state in F16 [30].
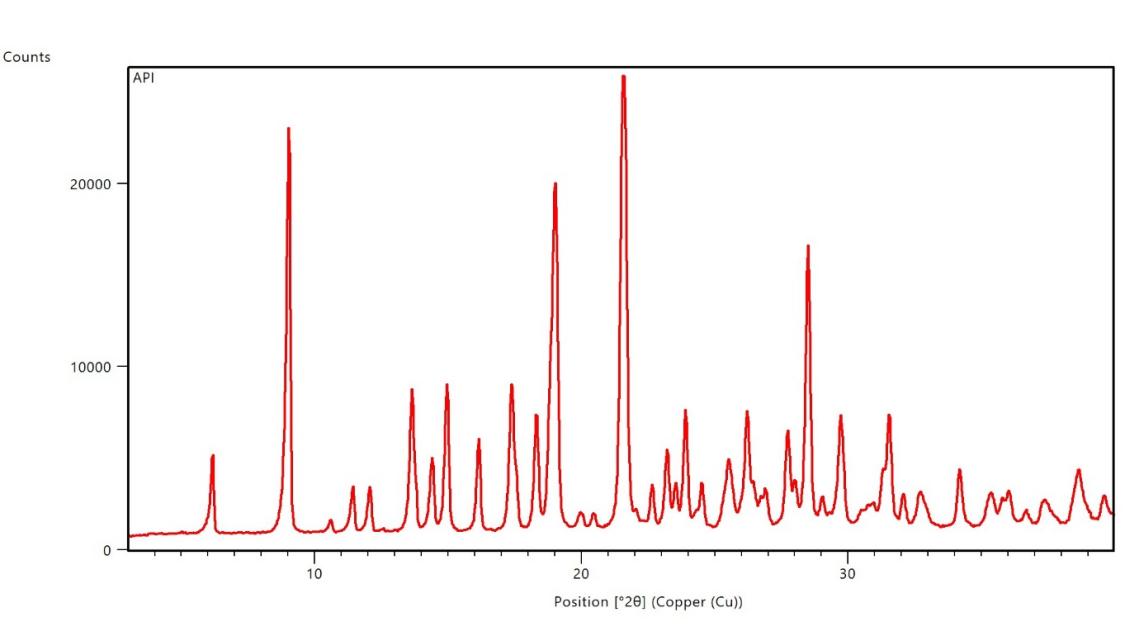
Fig. 3a: X-Ray powder diffraction image of the pure dolutegravir
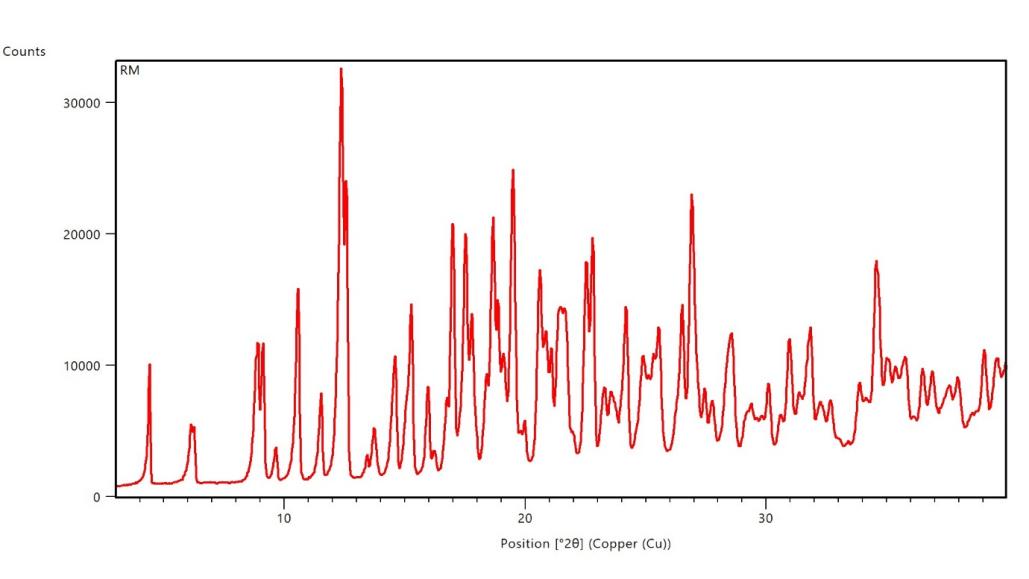
Fig. 3b: X-ray powder diffraction image of dolutegravir solid dispersion with Captisol®at a ratio of 1:4 w/w
Differential scanning calorimetry (DSC)
The DSC thermogram of pure Dolutegravir (fig. 4a) revealed a sharp endothermic peak at 242 °C, corresponding to its melting point, with no significant peaks observed below 242 °C. Formulation F16 (fig. 4b) showed a broad endothermic peak at 141.8 °C attributed to water loss, a glass transition temperature (Tg) at 130 °C, and a small endothermic peak at 200 °C likely due to carrier melting, with notably no Dolutegravir melting peak present. The pure drug exhibits a sharp melting peak at 242 °C, characteristic of its crystalline nature. Optimised solid dispersion (F16) shows the absence of the Dolutegravir melting peak, suggesting successful amorphisation of the drug [37].
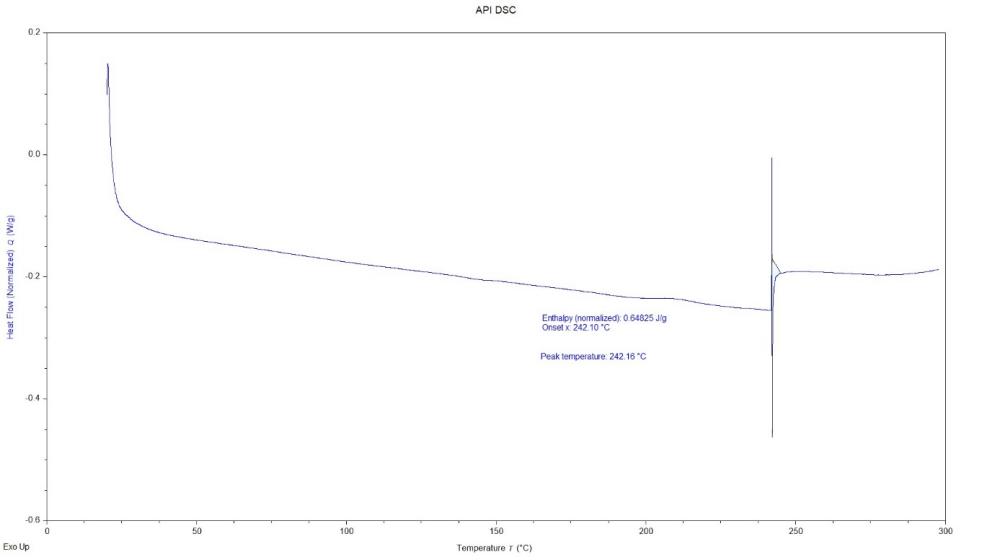
Fig. 4a: Differential scanning calorimetry image of the pure dolutegravir

Fig. 4b: Differential scanning calorimetry image of dolutegravir solid dispersion with Captisol®at a ratio of 1:4 w/w
FTIR
The most significant changes were observed in formulation F16 (fig. 5b), prepared by the fusion method. The N-H stretching peak of Dolutegravir shifted to 3300 cm-1 and became very broad. The C=O stretching moved to 1620 cm-1. Carrier peaks were highly prominent, with a broad O-H stretching peak from 3400-3100 cm-1. The aromatic C=C stretching peaks of dolutegravir (1580 cm-1 and 1510 cm-1) became less distinct and merged into a broader peak. The C-F stretching shifted to 1370 cm-1 [30].
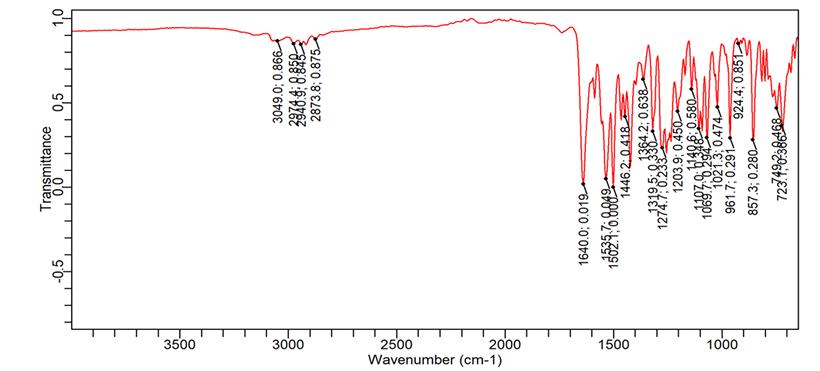
Fig. 5a: FTIR image of the pure dolutegravir
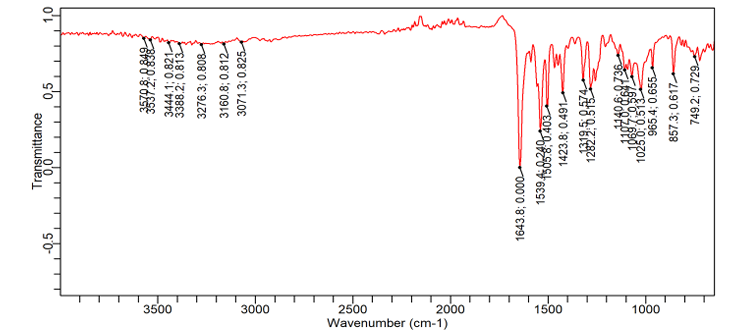
Fig. 5b: FTIR image of dolutegravir solid dispersion with Captisol® at a ratio of 1:4 w/w
Table 3: Percentage yield of solid dispersions
| Formulation code | Percentage yield (%) | Formulation code | Percentage yield (%) |
| F1 | 86.12±1.22 | F9 | 78.19±1.43 |
| F2 | 89.24±0.94 | F10 | 79.23±0.87 |
| F3 | 89.78±1.10 | F11 | 80.67±1.23 |
| F4 | 90.05±1.10 | F12 | 86.98±1.46 |
| F5 | 88.19±1.43 | F13 | 97.13±1.49 |
| F6 | 91.02±1.06 | F14 | 97.68±1.06 |
| F7 | 91.56±0.23 | F15 | 97.89±1.59 |
| F8 | 92.05±2.46 | F16 | 98.41±0.87% |
Data are given as mean±SD, (n=3)
Percentage yield
The percentage yield of prepared solid dispersions was calculated and reported in table 3. The highest percentage yield (98.41±0.87%) was found in solid dispersions F16 and lowest yield was (78.19±1.43) observed in F9. As seen in table 3.
In vitro dissolution studies of prepared solid dispersions
The in vitro dissolution study of dolutegravir solid dispersions revealed significant improvements in drug release compared to the pure drug. The pure drug exhibited a slow dissolution profile, with only 13.63% release after 60 min. In contrast, the formulations showed varying degrees of enhancement in dissolution rate. F16 demonstrated the most significant improvement among the formulations, achieving 79.14% drug release at 60 min. This represents a six-fold increase compared to the pure drug. F16 also showed rapid initial dissolution, with 12.9% release within the first 5 min, compared to only 0.93% for the pure drug. The dissolution profile of F16 exhibited a steady increase throughout the study period, indicating sustained and enhanced drug release. Other notable formulations include F15 and F14, achieving over 60% drug release at 60 min (62.14% and 61.53%, respectively). These formulations also showed rapid initial dissolution, with about 10% release in the first 5 min. Interestingly, formulations F1 to F13 showed moderate improvements in dissolution rate compared to the pure drug, but their performance was considerably lower than the top-performing formulations. The dissolution profiles of all formulations showed a general trend of rapid initial release followed by a more gradual increase over time. However, the extent and rate of release varied significantly among formulations. This variability could be attributed to differences in composition, preparation methods, or carrier ratios used in the solid dispersions. It is observed that formulations F14, F15, and F16 achieved over 50% drug release within 45 min, indicating their potential for rapid onset of action. The solid dispersions prepared by various methods showed marked improvements in dissolution rate compared to the pure drug. Among the preparation methods, the fusion method is the most effective technique, followed by the kneading method, co-grinding, and physical mixing. Formulations F13-F16 were prepared using the fusion method and consistently showed the highest dissolution rates. F16 exhibited the best performance with 79.14% drug release at 60 min, followed by F15 (62.21%), F14 (61.12%), and F13 (42.78%). This method proved superior in enhancing the dissolution rate of Dolutegravir [12]. Fig. 6 below shows in vitro dissolution profile of dolutegravir solid dispersion of various carriers.
Stability studies
The physical evaluation results of the optimized formulations are presented in table 4. Throughout the 6-month study period, the color of the solid dispersions remained consistent, appearing whitish to pale yellow at all time points [39]. This consistency in appearance suggests good physical stability of the formulations under the tested conditions. Assay results showed minimal degradation over the course of the study. The initial assay value was 99.5%. After 3 mo of storage at accelerated conditions, the assay value slightly decreased to 99.02%, indicating a minimal loss of 0.48%. At the 6-month time point, the assay value was 98.55%, representing a total decrease of 0.95% from the initial value. These results demonstrate good chemical stability of the dolutegravir in the solid dispersion formulations, with only minor degradation observed over the 6-month period under accelerated conditions.
Preparations and physicochemical evaluations of effervescent granules containing Dolutegravir solid dispersion
Physical evaluation
The Angle of repose of prepared granules of all formulations was found within the range of 24.645±0.06 to 33.7±0.01. The angle of repose results indicated that flow properties ranged from good to excellent. F1, F2, F3, F5, F6, F7 F9 and F10 showed excellent flow properties. F4, F11 and F12 showed good flow properties, whereas F8 showed fair flow properties, possibly due to the low concentration of Cetyl alcohol and sodium starch glycolate in the formulation. The concentration range of Cetyl alcohol in F13was 0.34%, and Sodium starch glycolate in F8 was 0.75%, which was less than other formulated preparations [40].
Bulk density was varied from 0.46±0.01 gm/ml to 0.82±0.01 gm/ml for 13 batches of formulation. And tapped density varied from 0.50±0.02 g/ml to 0.86±0.02 g/ml for 13 batches of formulations. Carr's compressibility index was calculated from the tapped density and bulk density. The values of Carr’s index of prepared granules were in the range of 4.61 to 17.39%. The Carr’s index results indicated flow properties ranged from fair to excellent. F1, F2, F3, F5, F6, F7, F9 and F10 showed excellent flow properties. F4, F11 and F12 showed good flow properties, whereas F8 and F13 showed fair flow properties, possibly due to the low concentration of sodium starch glycolate and Cetylalcohol [35].
The values of Hausner’s ratio of prepared granules were within the range of 1.04 to 1.20. Hausner’s ratio results indicated flow properties ranged from fair to excellent. F1, F2, F3, F5, F6, F7, F9 and F10 showed excellent flow properties. F4, F11 and F12 showed good flow properties, whereas F8 and F13 showed fair flow properties, possibly due to the low concentration of sodium starch glycolate and Cetyl alcohol. Table 4 contains the complete physical evaluation parameters of the formulated granules.
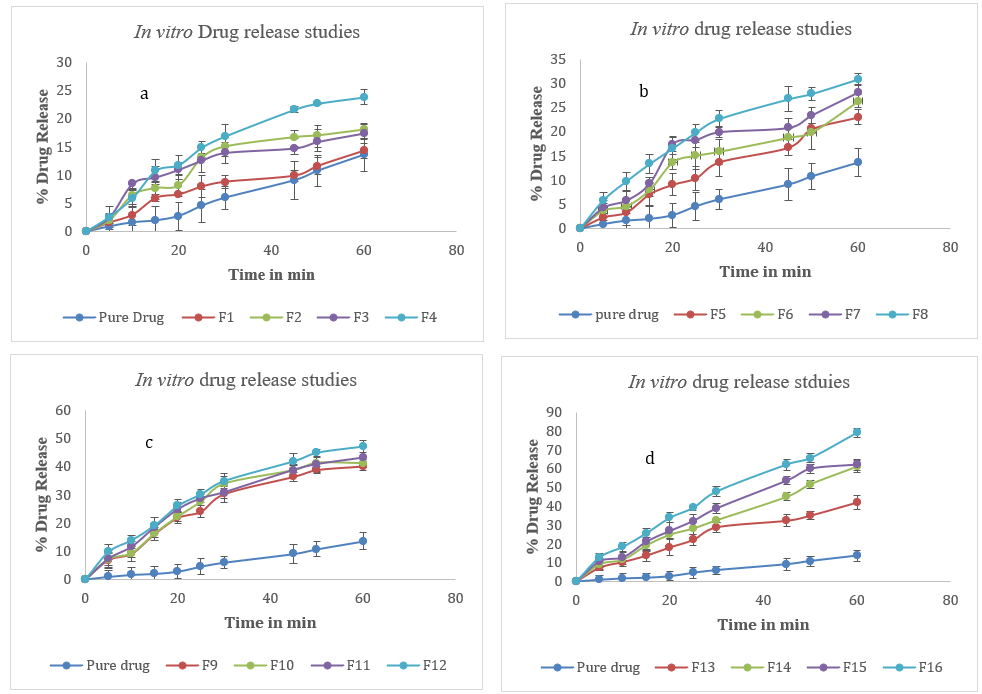
Fig. 6: In vitro drug release studies of dolutegravir solid dispersions a. F1-F4 b. F5-F8 c. F9-F12 d. F13-F16. Error bars indicate SD values
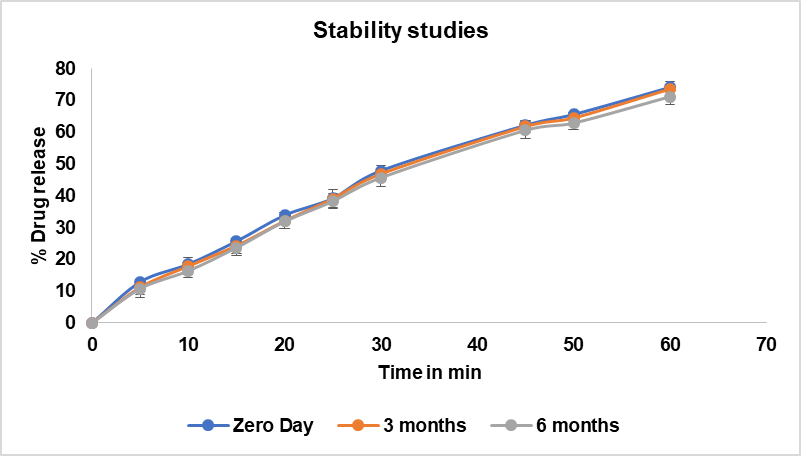
Fig. 7: Stability studies data of optimised formulation drug release during zero-month, 3rd month and 6th month. Error bars indicate SD
Drug content
All formulations show drug content values within the range of 90.2% to 99.5%, which is well within the typically accepted range of 90-110% for solid dispersions based effervescent granules as shown in table 3. The lowest drug content was observed in F7 (90.2%), while the highest was in F3 (99.5%) [30, 31]. This could be attributed to the more controlled nature of these preparation techniques, which may result in less drug loss during the formulation process.
Table 4: Result of physical evaluation of formulated granules
| FN | Bulk density | Tapped density | Hausner’s ratio | Carr’s CI | Angle of repose | Flow properties |
| 1 | 0.67±0.01 | 0.71±0.01 | 1.05 | 5.63% | 27.63±0.69 | Excellent |
| 2 | 0.82±0.01 | 0.86±0.02 | 1.04 | 4.65% | 28.42±0.42 | Excellent |
| 3 | 0.62±0.01 | 0.65±0.03 | 1.04 | 4.61% | 24.645±0.06 | Excellent |
| 4 | 0.66±0.02 | 0.77±0.01 | 1.16 | 14.28% | 33.335±0.007 | Good |
| 5 | 0.46±0.04 | 0.5±0.02 | 1.08 | 8% | 33.7±0.01 | Excellent |
| 6 | 0.46±0.04 | 0.5±0.02 | 1.08 | 8% | 33.7±0.01 | Excellent |
| 7 | 0.46±0.04 | 0.5±0.02 | 1.08 | 8% | 33.7±0.01 | Excellent |
| 8 | 0.57±0.04 | 0.68±0.04 | 1.19 | 16.17% | 27.145±0.007 | Fair |
| 9 | 0.46±0.01 | 0.5±0.02 | 1.08 | 8% | 33.7±0.01 | Excellent |
| 10 | 0.46±0.04 | 0.5±0.02 | 1.08 | 8% | 33.7±0.01 | Excellent |
| 11 | 0.62±0.02 | 0.73±0.04 | 1.17 | 15.06% | 25.765±0.04 | Good |
| 12 | 0.52±0.02 | 0.6±0.02 | 1.15 | 13.34% | 26.7±0.59 | Good |
Data are given as mean±SD, (n=3)
Effervescence cessation time
The values of effervescence cessation time were in the range of 19.5.5±2.12 to 81.5±2.12 seconds. Among all the 13 formulations F5, F6, F7, F9 and F10 showed least effervescent time i. e., 19.5±2.12.
Compared with other formulations, F2 showed the highest effervescent time, i. e., 81.5±2.12, which might be due to the higher concentration of Cetyl alcohol and sodium starch glycolate than in other formulations. The concentration of Cetyl alcohol and sodium starch glycolate was 10% and 8%, respectively, hence the longer effervescence time [34].
Table 5: Effervescence cessation time of formulated granules
| Formulation code | Drug content %(w/w) | Amount of drug released % | Effervescent time (sec) |
| F1 | 93.9±0.14 | 90.2±0.13 | 43.5±0.70 |
| F2 | 95.6±0.13 | 91.4±0.12 | 81.5±2.12 |
| F3 | 99.5±0.11 | 99.6 ±0.18 | 19.5±2.12 |
| F4 | 95.1±0.99 | 95.3±0.11 | 22.5±3.53 |
| F5 | 90.6±0.14 | 91.1±0.23 | 24.5±0.70 |
| F6 | 92.1±0.12 | 90.6±0.14 | 19.5±2.12 |
| F7 | 90.2±0.11 | 93.9±0.12 | 19.5±2.12 |
| F8 | 93.9±0.13 | 90.6±0.14 | 29±1.414 |
| F9 | 92.2±0.55 | 93.9±0.56 | 19.5±2.12 |
| F10 | 96.6±0.1 | 89.5±0.32 | 19.5±2.12 |
| F11 | 95.1±0.4 | 91.2±0.11 | 45.5±2.12 |
| F12 | 95.6±0.13 | 90.2±0.12 | 60.5±0.70 |
*Results are expressed as a mean±SD, n=3.
In vitro dissolution study of effervescent granules
The drug release studies are conducted in pH 1.2 acidic buffer to know about the drug release patterns of the prepared effervescent granules. The % amount of drug released after 5 min obtained for the 12 formulations of effervescent granules of dolutegravir is presented in (table 5). The results show that all 12 formulations had a good release profile within 5 min. The improvement of effervescent granules dissolution occurs due to the bursting of the granules into minute particles, which is facilitated by effervescence production. F3 shows the highest percentage of drug released, 99.5% within 5 min. The good release profile of F3 may be attributed to the combination of cetyl alcohol and sodium starch glycolate [36].
CONCLUSION
This present work successfully developed and fully characterized the effervescent granules containing dolutegravir solid dispersion for special subjects such as children and the elder, with the purpose of solubility and bitterness enhancement, along with stomach acidic protection. The optimal solid dispersion with Captisol®at a drug: polymer ratio of 1:4 (w/w), prepared by the fusion method, significantly increased the Dolutegravir solubility to nearly 4 times. Analytically, the solid dispersion possessed intermolecular bonding between the drug and polymer, transforming the drug from a crystalline to an amorphous state. Finally, a total of 12 formulations of the effervescent granules containing the solid dispersion were developed using various excipients such as sweeteners, gas generators, pH modulators, and glidants/lubricants. The results show that all 12 formulations had a good release profile within 5 min. F3 shows the highest drug content (99.5±0.11), the highest percentage of drug release 99.5% within 5 min and the effervescent cessation time 19.5±2.12 sec. The good release profile of F3 may be attributed to the combination of cetyl alcohol and sodium starch glycolate, indicating that the final product is suitable to be further research in in vivo and in clinical settings to become a dosage form containing Dolutegravir with high bioavailability that is appropriate for children and the elder.
ACKNOWLEDGEMENT
The authors would like to thank Joginpally B. R. Pharmacy College for providing facilities to conduct the research and NIPER, Hyderabad, for their support in conducting DSC, SEM, FTIR and XRD characterisation process.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
Gottemukkula Lakshmi Devi designed the experiment, Aishwarya, M. Jyothiraditya, B. Manikanth, K. Shivakrishna and P. Sainath Goud formulated the formulations, and analysed the results. Sunitha Sampathi reviewed the manuscript and gave valuable suggestions to complete this research work.
CONFLICT OF INTERESTS
The authors declare no conflict of interest
REFERENCES
Sarode IM, Jindal AB. Current status of dolutegravir delivery systems for the treatment of HIV-1 infection. J Drug Deliv Sci Technol. 2022 Oct;76:103802. doi: 10.1016/j.jddst.2022.103802.
Smith SJ, Zhao XZ, Passos DO, Lyumkis D, Burke TR, Hughes SH. Integrase strand transfer inhibitors are effective anti-HIV drugs. Viruses. 2021 Feb 1;13(2):205. doi: 10.3390/v13020205, PMID 33572956.
Development of practical synthetic method for the preparation of dolutegravir sodium, a potent HIV-1 integrase inhibitor for the treatment of HIV infectious disease. Yukigoseikyo Kaishi. 2023 Mar 7.
Muse D, Tarau E, Lefeber C, Sohns M, Brett M, Goldberg J. Pharmacokinetics, safety, and efficacy of tapentadol oral solution for treating moderate to severe pain in pediatric patients. J Pain Res. 2019;12:1777-90. doi: 10.2147/JPR.S197039, PMID 31213888.
Galande AD, Khurana NA, Mutalik S. Pediatric dosage formschallenges and recent developments: a critical review. J Appl Pharm Sci. 2020 Jul;10(7):155-66. doi: 10.7324/JAPS.2020.10718.
Dunne J, Rodriguez WJ, Murphy MD, Beasley BN, Burckart GJ, Filie JD, et al. Extrapolation of adult data and other data in pediatric drug-development programs. Pediatrics. 2011 Nov;128(5):e1242-9. doi: 10.1542/peds.2010-3487, PMID 22025597.
Bassat Q. The unmet needs of paediatric therapeutics in poor countries. J Trop Pediatr. 2015 Dec 1;61(6):403-6. doi: 10.1093/tropej/fmv081, PMID 26589503.
Brown A, Rice SM, Rickwood DJ, Parker AG. Systematic review of barriers and facilitators to accessing and engaging with mental health care among at-risk young people. Asia Pac Psychiatry. 2016;8(1):3-22. doi: 10.1111/appy.12199, PMID 26238088.
Nikam VK, Shete SK, Khapare JP. Most promising solid dispersion technique of oral dispersible tablet. Beni Suef Univ J Basic Appl Sci. 2020;9(1). doi: 10.1186/s43088-020-00086-4.
Wasilewska K, Winnicka K. Ethylcellulose-a pharmaceutical excipient with multidirectional application in drug dosage forms development. Materials (Basel). 2019 Oct;12(20):3386. doi: 10.3390/ma12203386, PMID 31627271.
Gottemukkula LD, Pathuri R. Development and optimization of a dolutegravir nanosuspension using box behnken design. Int J App Pharm. 2024 May;16(3):129-39. doi: 10.22159/ijap.2024v16i3.50315.
Malkawi R, Malkawi WI, Al-Mahmoud Y, Tawalbeh J. Current trends on solid dispersions: past, present, and future. Adv Pharmacol Pharm Sci. 2022;2022:5916013. doi: 10.1155/2022/5916013, PMID 36317015.
Bhairam M, Shukla SS, Gidwani B, Pandey RK. Solid dispersion of dolutegravir: formulation development, characterization, and pharmacokinetic assessment. Int J Pharm Qual Assur. 2022 Oct;13(4):496-503. doi: 10.25258/ijpqa.13.4.24.
Sapkal SB, Adhao VS, Thenge RR, Darakhe RA, Shinde SA, Shrikhande VN. Formulation and characterization of solid dispersions of etoricoxib using natural polymers. Turk J Pharm Sci. 2020;17(1):7-19. doi: 10.4274/tjps.galenos.2018.04880, PMID 32454755.
Mupparaju S, Suryadevara V, Yallam S, Doppalapudi S, Reddyvallam LS, Anne R. Formulation and evaluation of dolutegravir sodium solid dispersions and fast dissolving tablets using poloxamer-188 and jackfruit seed starch as excipients. Asian J Pharm Clin Res. 2019 Apr;12(6):181-90. doi: 10.22159/ajpcr.2019.v12i6.33302.
Bhairam M, Pandey RK, Shukla SS, Gidwani B. Preparation, optimization, and evaluation of dolutegravir nanosuspension: in vitro and in vivo characterization. J Pharm Innov. 2023;18(4):1798-811. doi: 10.1007/s12247-023-09756-z.
Raj H, Sharma A, Sharma S, Verma KK, Chaudhary A. Mucoadhesive microspheres: a targeted drug delivery system. J Drug Delivery Ther. 2021 Apr;11(2-S):150-5. doi: 10.22270/jddt.v11i2-S.4791.
Nikghalb LA, Singh G, Singh G, Kahkeshan KF. Solid dispersion: methods and polymers to increase the solubility of poorly soluble drugs. J Appl Pharm Sci. 2012 Oct;2(10):170-5. doi: 10.7324/JAPS.2012.21031.
Jadav NB, Paradkar A. Solid dispersions. In: Grumezescu AM, editor. Nanopharmaceuticals. Vol. 1. Amsterdam: Elsevier; 2020. p. 91-120. doi: 10.1016/B978-0-12-817778-5.00005-1.
Daravath B, Tadikonda RR, Vemula SK. Formulation and pharmacokinetics of gelucire solid dispersions of flurbiprofen. Drug Dev Ind Pharm. 2015 Aug;41(8):1254-62. doi: 10.3109/03639045.2014.940963, PMID 25039470.
Prasad R, Radhakrishnan P, Singh SK, Verma PR. Furosemide-Soluplus® solid dispersion: development and characterization. Recent Pat Drug Deliv Formul. 2017 Apr;11(3):211-20. doi: 10.2174/1872211311666171129120020, PMID 29189186.
Pathuri R, Gottemukkula LD. Development and optimization of a self-nanoemulsifying drug delivery system (SNEDDS) for enhanced oral delivery of dolutegravir. J Pharm Innov. 2025 Apr;20(2). doi: 10.1007/s12247-025-09951-0.
Pandi P, Bulusu R, Kommineni N, Khan W, Singh M. Amorphous solid dispersions: an update for preparation, characterization, mechanism on bioavailability, stability, regulatory considerations and marketed products. Int J Pharm. 2020 Aug;586:119560. doi: 10.1016/j.ijpharm.2020.119560, PMID 32565285.
Sharma A, Jain CP, Tanwar YS. Preparation and characterization of solid dispersions of carvedilol with Poloxamer 188. J Chil Chem Soc. 2013;58(1):1553-7. doi: 10.4067/S0717-97072013000100012.
Li X, Peng H, Tian B, Gou J, Yao Q, Tao X. Preparation and characterization of azithromycin – Aerosil 200 solid dispersions with enhanced physical stability. Int J Pharm. 2015 May;486(1-2):175-84. doi: 10.1016/j.ijpharm.2015.03.029, PMID 25794608.
Gottemukkula LD, Pathuri R, Sampathi S. Dolutegravir solid dispersions as oro-dispersible tablets: to ameliorate the integrase inhibition effect. Ind J Pharm Edu Res. 2025;59(3s):s863-73. doi: 10.5530/ijper.20255690.
Kim MJ, Lee JH, Yoon H, Kim SJ, Jeon DY, Jang JE. Preparation, characterization and in vitro dissolution of aceclofenac-loaded PVP solid dispersions prepared by spray drying or rotary evaporation method. J Pharm Investig. 2013 Apr;43(2):107-13. doi: 10.1007/s40005-013-0058-3.
Khan AW, Kotta S, Ansari SH, Sharma RK, Ali J. Enhanced dissolution and bioavailability of grapefruit flavonoid naringenin by solid dispersion utilizing fourth generation carrier. Drug Dev Ind Pharm. 2015 May;41(5):772-9. doi: 10.3109/03639045.2014.902466, PMID 24669978.
Yadav G, Kumar P, Kumar Y, Singh PK. Dolutegravir, second generation integrase inhibitor: A New Hope for HIV patient. European Journal of Molecular and Clinical Medicine. 2018;5(1):20-9. doi: 10.5334/ejmcm.252.
Al-Mousawy J, Al-Hussainy Z, Alaayedi M. Formulation and evaluation of effervescent granules of ibuprofen. Int J App Pharm. 2019 Nov;11(6):66-9. doi: 10.22159/ijap.2019v11i6.34912.
Huynh DT, Hai HT, Hau NM, Lan HK, Vinh TP, Tran VD. Preparations and characterizations of effervescent granules containing azithromycin solid dispersion for children and elder: solubility enhancement, taste-masking, and digestive acidic protection. Heliyon. 2023 Jun;9(6):e16592. doi: 10.1016/j.heliyon.2023.e16592, PMID 37292293.
Faisal A. Formulation by design approach for effervescent granules of vitamin C using statistical optimization methodologies. JAPR. 2020;8(4):62-9. doi: 10.18231/j.joapr.2020.v.8.i.4.62.69.
Basha M, Salama A, Noshi SH. Soluplus® based solid dispersion as fast disintegrating tablets: a combined experimental approach for enhancing the dissolution and antiulcer efficacy of famotidine. Drug Dev Ind Pharm. 2020 Feb;46(2):253-63. doi: 10.1080/03639045.2020.1716376, PMID 31937139.
Reese MJ, Savina PM, Generaux GT, Tracey H, Humphreys JE, Kanaoka E. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab Dispos. 2013 Feb;41(2):353-61. doi: 10.1124/dmd.112.048918, PMID 23132334.
Yarlagadda DL, Nayak AM, Brahmam B, Bhat K. Exploring the solubility and bioavailability of sodium salt and its free acid solid dispersions of dolutegravir. Adv Pharmacol Pharm Sci. 2023;2023:7198674. doi: 10.1155/2023/7198674, PMID 37383518.
Dos Santos C, Buera MP, Mazzobre MF. Phase solubility studies of terpineol with β-cyclodextrins and stability of the freeze-dried inclusion complex. Procedia Food Sci. 2011;1:355-62. doi: 10.1016/j.profoo.2011.09.055.
Waghchoure K. A review on: effervescent tablet. Int J Pharm Res Appl. 2021;8(1):1246-55. doi: 10.35629/7781-080112461255.