
Int J App Pharm, Vol 18, Issue 1, 2026, 162-174Original Article
IN SILICO STUDY OF NEW ACETAZOLAMIDE ANALOGUES BEARING THIAZOLE MOIETY WITH PROMISING CARBONIC ANHYDRASE INHIBITORY ACTIVITY
HUSSEIN I. ABDULHUSSEIN1*, NOOR H. NASER2
1Department of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Kufa, Najaf, Iraq. 2Department of Pharmaceutical Chemistry, College of Pharmacy, Al-Zahraa University for Women, Karbala, Iraq
*Corresponding author: Hussein I. Abdulhussein; *Email: husseini.almahmud@student.uokufa.edu.iq
Received: 08 Jul 2025, Revised and Accepted: 28 Oct 2025
ABSTRACT
Objective: Cancer is the second leading cause of death worldwide. Multiple targets can be hit for cancer treatment; carbonic anhydrase enzyme XII is one of these targets because it plays a vital role in the tumour microenvironment.
Methods: ADMETlab 3.0 platform used to predict (absorption, distribution, metabolism, excretion, and toxicity) characteristics, while molecular docking managed by the molecular operating environment program MOE 2015.1.
Results: The molecular docking analysis revealed that all nineteen designed compounds exhibited favourable binding affinities toward carbonic anhydrase enzymes XII (PDB ID: 1JD0) and IX (PDB ID: 3IAI), and most of the designed compounds satisfy the predominant drug-likeness requirements.
Conclusion: Acetazolamide's binding affinity for the carbonic anhydrase enzyme can be enhanced by including a substituted thiazole ring.
Keywords: Cancer, Docking, Carbonic anhydrase, Acetazolamide, Thiazole
© 2026 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2026v18i1.55957 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Cancer is a term used to describe several diseases distinguished by an abnormal proliferation of defective cells that can invade and damage normally functioning tissues [1]. More than 90% of cancer-related fatalities are attributable to metastasis, which nearly always has a fatal outcome [2]. The prevalence of cancer is increasing and has grown into the second major cause of fatality internationally [3-5]. Nearly 20 million new cases of cancer were reported in 2022, with 9.7 million deaths attributable to the disease, according to the International Agency for Research on Cancer (IARC) [6]. Chemicals such as nicotine, radiation, infectious agents, genetic mutations, hormone imbalances, and immune system abnormalities are just some of the inside-out variables contributing to the occurrence of cancer [7]. Cancer's prognosis fluctuates by numerous characteristics, including gender, age, and sex [8].
Hypoxia, featuring decreased oxygen levels, is a distinctive characteristic of the tumor microenvironment in all solid malignancies [9]. Cancer is ultimately lethal when the disease becomes resistant to therapy and spreads to other parts of the body [10]. The search for novel small-molecule chemotherapeutic agents that are both efficacious and safe for cancer treatment or prevention has heightened in recent years due to multidrug resistance [11].
Carbonic anhydrases are metalloenzymes having a fundamental part in the catalysis of the bidirectional CO2 hydration process (CO2+H2O ⇄ HCO3-+H+), enabling them to regulate the levels of CO2, HCO3-, and H+ [12, 13]. Since human carbonic anhydrase (hCA), and notably hCA IX and hCA XII isoforms, participate in critical roles in cancer cell metastasis and hypoxia response and are expressed mainly by cancer cells, they have attracted the interest of numerous researchers involved in the development of drugs that target cancer [14]. Multiple studies highlight the overexpression of carbonic anhydrase XII in cancer, one of these studies is ‘High expression of carbonic anhydrase 12 (CA12) is associated with good prognosis in breast cancer’, which published in 2019 [15]. Other studies show the strong involvement of carbonic anhydrase IX and cancer, such as ‘Carbonic anhydrase IX (CA9) modulates tumor-associated cell migration and invasion’ that published in 2011 [16]. Carbonic anhydrase inhibitors fall into one of four categories based on how they attach to the enzyme's active site. The most significant group is zinc ion binders, to which sulfonamide belongs [17].
Sulfonamides demonstrate many pharmacological activities, including the anti-carbonic anhydrase effect, enabling their application in treating diverse conditions such as diuresis, hypoglycemia, thyroiditis, inflammation, and glaucoma [18]. Acetazolamide is a longstanding medication employed for several purposes. The medication is infrequently utilized, primarily because of perceived inadequate efficacy and undesirable effects. Acetazolamide functions as a noncompetitive inhibitor of carbonic anhydrase, which exists in many subtypes in humans. Acetazolamide induces acidification in both intracellular and extracellular environments, hence activating acid-sensing ion channels [19].
Thiazole is a sulfur-nitrogen-containing heterocyclic moiety that plays a significant role in medicinal chemistry. It is a fundamental building block of numerous molecules with medical importance, both naturally occurring and artificially produced [20]. The antiparasitic, antifungal, antibacterial, and antiproliferative biological effects of thiazole compounds are well-documented [21].
This study aims to design new acetazolamide-thiazole-based compounds with carbonic anhydrase inhibitory activity.
MATERIALS AND METHODS
Study design
Considering the preceding introduction, new acetazolamide derivatives bearing a thiazole ring moiety were constructed to function as a breast cancer treatment by inhibiting the carbonic anhydrase enzyme. The synthesis process of the designed acetazolamide derivatives is illustrated in scheme 1.
ADMET and drug-likeness properties
Research and development limitations in terms of efficacy and safety are often attributed to issues with ADME (absorption, distribution, metabolism, and excretion) features, with different levels of toxicity. Drug development failures can be significantly reduced with the help of quick ADMET evaluation. In this study, ADMETlab 3.0, a complementary web-based platform designed under the umbrella of the CBDD group of Xiangya School of Pharmaceutical Sciences, Central South University, has been utilized. ADMETlab 3.0 provides better performance than accessible platforms such as SwissADME and offers additional drug-likeness, physicochemical, and ADMET characteristics. To make ADMET predictions, the designed compounds were loaded into the ADMETlab 3.0 as SMILES [22-24]. The prediction models of ADMETlab 3.0 are constructed using the Directed Message Passing Neural Network (DMPNN) architecture. These are multi-task deep learning algorithms that analyze molecular graphs by transmitting messages along bonds to acquire structural information. Furthermore, these graph-derived representations are integrated with molecular descriptors, enhancing both resilience and predictive precision [25].
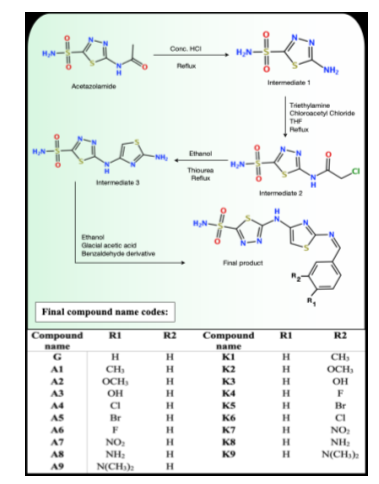
Scheme 1: Chemical synthesis of the designed compounds
Ligand and receptor preparation and molecular docking
The molecular docking study was conducted using the Molecular Operating Environment program MOE 2015.1 to elucidate the interaction of the designed compounds with human carbonic anhydrase enzyme XII and human carbonic anhydrase enzyme IX [26, 27].
Docking validation
The accuracy and reliability of the docking process were ensured by validation through RMSD calculation via re-docking of the co-crystalized ligand; the validation RMSD result was 1.2561, shown in fig. 1, which is an acceptable value.
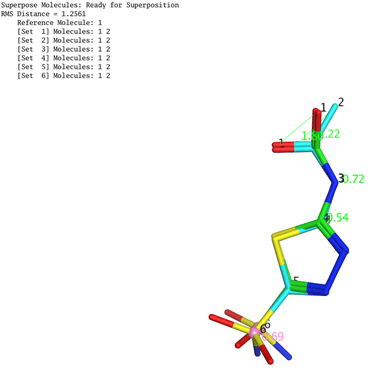
Fig. 1: Molecular docking validation result
Ligand preparation
The designed compound two dimension 2D structures were drawn using the ChemDraw Professional 12.0 program, and then these drawn structures were set up for docking by MOE 2015.1 using the following steps: structure protonation to add the necessary hydrogen atoms to the designed compound 2D structures, partial charge addition, and energy minimization (forcefield used in ligand energy minimization is MMFF94x, this field used only in this part of docking then changed to the MOE default forcefield Amber10:EHT), as shown in fig. 2 [28].
Receptor preparation
The human carbonic anhydrase crystal structures were downloaded from pdb (protein data bank, https://www.rcsb.org) in the form of PDB format, these crystal structures have the pdb code 1JD0 and 3IAI, and then prepared for docking using MOE 2015.1 in multiple steps, the first step involve the removal of solvent, unnecessary parts of the enzyme, and ligand that attached to the original enzyme site, the following steps involve structural protonation to add the necessary hydrogen atoms to the hCA crystal structure, correction of structural issues, fixing the charge, and lastly identifying the binding pocket and creating a dummy for it, the process is explained in fig. 2 [29].
Docking procedure
The docking study proceeded using the prepared hCA site and the prepared ligands. Each ligand molecule was illustrated in 5 different poses, for a total of 50 (this is a pure automated process). All steps are listed in fig. 2. This study utilized hCA XII, identified by pdb code 1JD0, and hCA IX, identified by pdb code 3IAI, together with the designed compounds. The reference utilized in this study is acetazolamide, as it serves as the foundational structure for all the designed compounds. The positive control structure used is SLC-0111, recognized for its established binding affinity to hCA XII and hCA IX. The employed forcefield is Amber10:EHT, and the dimensions of the cell shape are 90 x 90 x 90 (space group P1) [30, 31].
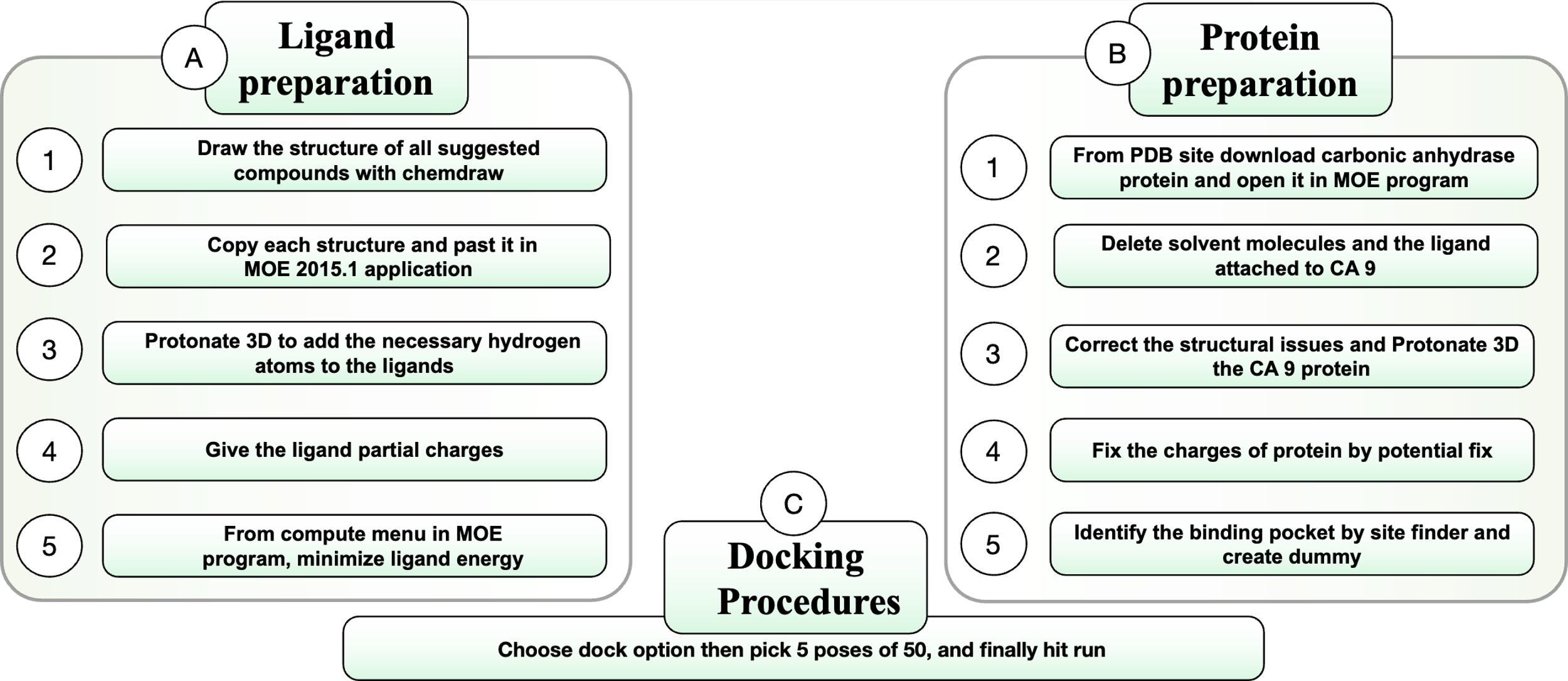
Fig. 2: Molecular docking study steps using molecular operating environment program
Table 1: Predicted physicochemical characteristics of the designed compounds
| Compound name | M. Wt | nHD | nHA | LogP | TPSA | nRot | LogS | LogD7.4 |
| G | 366 | 3 | 8 | 1.742 | 123.22 | 5 | -4.154 | 2.108 |
| A1 | 380.02 | 3 | 8 | 2.258 | 123.22 | 5 | -4.42 | 2.553 |
| A2 | 396 | 3 | 9 | 1.839 | 132.45 | 6 | -4.112 | 2.161 |
| A3 | 382 | 4 | 9 | 1.335 | 143.45 | 5 | -3.283 | 1.597 |
| A4 | 399.96 | 3 | 8 | 2.285 | 123.22 | 5 | -4.329 | 2.559 |
| A5 | 443.91 | 3 | 8 | 2.283 | 123.22 | 5 | -4.379 | 2.596 |
| A6 | 383.99 | 3 | 8 | 2.149 | 123.22 | 5 | -4.249 | 2.479 |
| A7 | 410.99 | 3 | 11 | 1.454 | 166.36 | 6 | -4.032 | 1.871 |
| A8 | 381.01 | 5 | 9 | 0.797 | 149.24 | 5 | -3.259 | 1.141 |
| A9 | 409.04 | 3 | 9 | 1.606 | 126.46 | 6 | -3.798 | 2.015 |
| K1 | 380.02 | 3 | 8 | 2.129 | 123.22 | 5 | -4.341 | 2.444 |
| K2 | 396.01 | 3 | 9 | 1.869 | 132.45 | 6 | -4.154 | 2.205 |
| K3 | 382 | 4 | 9 | 1.29 | 143.45 | 5 | -3.233 | 1.554 |
| K4 | 399.96 | 3 | 8 | 2.336 | 123.22 | 5 | -4.47 | 2.6 |
| K5 | 443.91 | 3 | 8 | 2.368 | 123.22 | 5 | -4.529 | 2.628 |
| K6 | 383.99 | 3 | 8 | 2.152 | 123.22 | 5 | -4.305 | 2.477 |
| K7 | 410.99 | 3 | 11 | 1.51 | 166.36 | 6 | -4.135 | 1.916 |
| K8 | 381.01 | 5 | 9 | 0.846 | 149.24 | 5 | -3.241 | 1.194 |
| K9 | 409.04 | 3 | 9 | 1.741 | 126.46 | 6 | -3.955 | 2.12 |
M. Wt, Molecular weight; nHA, no. of hydrogen bond acceptors; nHD, no. of hydrogen bond donors; nRot, no. of rotatable bonds; TPSA, total polar surface area; logS, aqueous solubility; logP, octanol-water partition coefficient; logD7.4, distribution coefficient.
RESULTS AND DISCUSSION
ADMET and drug-likeness properties
Physicochemical parameters
From a physicochemical standpoint, it was noted that many of the designed compounds meet the most popular drug-likeness criteria, which are Lipinski's rule of five (RO5) and Verber's rule. An essential component of oral absorption is the logarithm of aqueous solubility, abbreviated as LogS. The acceptable range for LogS is between-4 and 0.5 log mol/l. The solubility of A3, A8, A9, K3, K8, and K9 is within the permitted range, according to the table 1, whereas the LogS values of the other molecules named G, A1, A2, A4, A5, A6, A7, K1, K2, K4, K5, K6, K7 are outside that range reflecting a low water solubility. Water solubility can be enhanced by complexing these compounds with metals. At a pH of 7.4, the logarithm of the n-octanol/water distribution coefficients is represented by LogD. In order to dissolve in bodily fluids and pass through the biomembrane, potential pharmaceuticals must exhibit a balance between lipophilicity and hydrophilicity. As a result, early-stage drug research candidate compounds need to have appropriate logD7.4 values, which range from 1 to 3 mol/l. The logD7.4 values of all the chemicals that were the focus of this study were found to be within the acceptable range. That being said, some of the designed compounds as A3, A8, A9, K3, K8, and K9 exhibit well-respected physicochemical properties, which make them promising candidates for intestinal absorption [32]. Fig. 3 shows a radar-like image of each designed compound to explain the physicochemical properties line of each compound (in yellow colour) with the acceptable range marketed with blue colour.

Fig. 3: The predicted physicochemical properties of the designed compounds
Predicted absorption parameters
The oral administration of drugs is the preferred method due to its convenience and safety. Water solubility and permeability across the biological membrane of intestinal cells (intestinal permeability) are the two fundamental characteristics of oral medications. Consequently, assessing the intestinal permeability of novel compounds is essential in drug discovery efforts to prevent late-stage attrition. ADMET Lab 3.0 contains many algorithms for evaluating drug absorption parameters. The Caco-2 (Caucasian colon adenocarcinoma cell lines permeability), PMPA (Parallel artificial membrane permeability assay), and MDCK (Madin–Darby canine kidney permeability) are utilized to evaluate drug permeability in vitro. All designed compounds investigated in this study demonstrated superior parallel artificial membrane permeability assay (PAMPA) but exhibited inadequate MDCK and Caco-2 permeability. The designed compounds are anticipated not to be inhibitors nor substrates of Para Glycoprotein (P. gp) (table 2). This suggests that the absorption mechanism of these chemicals is likely by passive diffusion rather than active uptake. Moreover, all compounds exhibited an exceptional anticipated value for human intestinal absorption (HIA), a critical measure for assessing oral medication bioavailability (F). Correspondingly, designed compounds exhibited a good F50 (Molecules having a bioavailability ≥ 50%) (table 2). In summary, all compounds demonstrated limited absorption properties due to poor Caco-2 and MDCK models, indicating their limited potential viability as orally administrable medicines [33].
Table 2: Predicted absorption characteristics of the designed compounds
| Compound name | Caco-2 | PAMPA | MDCK | P. gp inhibition | P. gp substrate | HIA | F50% |
| G | poor | good | poor | no | no | good | good |
| A1 | poor | good | poor | no | no | good | good |
| A2 | poor | good | poor | no | no | good | good |
| A3 | poor | good | poor | no | no | good | good |
| A4 | poor | good | poor | no | no | good | good |
| A5 | poor | good | poor | no | no | good | good |
| A6 | poor | good | poor | no | no | good | good |
| A7 | poor | good | poor | no | no | good | good |
| A8 | poor | good | poor | no | no | good | good |
| A9 | poor | good | poor | no | no | good | good |
| K1 | poor | good | poor | no | no | good | good |
| K2 | poor | good | poor | no | no | good | good |
| K3 | poor | good | poor | no | no | good | good |
| K4 | poor | good | poor | no | no | good | good |
| K5 | poor | good | poor | no | no | good | good |
| K6 | poor | good | poor | no | no | good | good |
| K7 | poor | good | poor | no | no | good | good |
| K8 | poor | good | poor | no | no | good | good |
| K9 | poor | good | poor | no | no | good | good |
Caco-2, Caucasian colon adenocarcinoma cell lines permeability; PMPA, Parallel artificial membrane permeability assay, MDCK, Madin–Darby canine kidney permeability; P. gp, para-glycoprotein; HIA, human intestinal absorption; F50, bioavailability ≥ 50%.
Table 3: Predicted distribution characteristics of the designed compounds
| Compound name | PPB% | Vdss | BBB | OATP1B1 inhibitor | OATP1B3 inhibitor | BCRP inhibitor | MRP1 inhibitor |
| G | 97.203 | -0.15 | 0.01 | 0.995 | 1 | 0 | 0.902 |
| A1 | 97.725 | -0.153 | 0.001 | 0.998 | 1 | 0 | 0.671 |
| A2 | 96.749 | -0.246 | 0.001 | 0.999 | 1 | 0 | 0.204 |
| A3 | 96.017 | -0.306 | 0 | 0.998 | 1 | 0 | 0.932 |
| A4 | 98.009 | -0.202 | 0.004 | 0.997 | 1 | 0 | 0.891 |
| A5 | 97.864 | -0.08 | 0.008 | 0.998 | 1 | 0 | 0.941 |
| A6 | 97.287 | -0.17 | 0.002 | 0.997 | 1 | 0 | 0.784 |
| A7 | 96.842 | -0.295 | 0 | 0.987 | 0.999 | 0 | 0.999 |
| A8 | 95.658 | -0.279 | 0.003 | 0.987 | 0.998 | 0 | 0.906 |
| A9 | 97.194 | -0.105 | 0 | 0.999 | 1 | 0 | 0.775 |
| K1 | 97.956 | -0.115 | 0.01 | 0.997 | 0.999 | 0 | 0.964 |
| K2 | 97.38 | -0.178 | 0.002 | 0.997 | 1 | 0 | 0.94 |
| K3 | 95.698 | -0.364 | 0.003 | 0.996 | 1 | 0 | 0.934 |
| K4 | 98.051 | -0.124 | 0.035 | 0.99 | 0.998 | 0 | 0.957 |
| K5 | 98.066 | -0.014 | 0.053 | 0.993 | 0.999 | 0 | 0.961 |
| K6 | 97.591 | -0.111 | 0.01 | 0.989 | 0.998 | 0 | 0.954 |
| K7 | 97.473 | -0.247 | 0 | 0.969 | 0.996 | 0 | 0.999 |
| K8 | 95.863 | -0.278 | 0.01 | 0.994 | 0.999 | 0 | 0.923 |
| K9 | 97.342 | -0.092 | 0.002 | 0.998 | 0.999 | 0 | 0.956 |
Plasma Protein Binding (PPB), Volume of distribution at steady state (Vdss), blood-brain barrier (BBB) permeability, Breast Cancer Resistance Protein (BCRP), Organic Anion Transporting Polypeptide 1B1 (OATP1B1), 1B3 (OATP1B3), Multidrug Resistance Protein 1 (MRP1).
Predicted distribution parameters
Plasma Protein Binding (PPB), Volume of Distribution at Steady State (Vdss), blood-brain barrier (BBB) penetration, and specific transporter inhibition profiles were considered to evaluate the distribution parameters of the designed compounds. All designed compounds had a predicted PPB exceeding 90%, indicating a significantly reduced free drug fraction available for target engagement that limits the ability of these designed compounds to reach their target, hCA XII, which is an intracellular enzyme. An examination of the medication database has disclosed that a substantial majority of small drug molecules demonstrate a PPB percentage beyond 90%. Moreover, compounds exhibiting acidity and a logD7.4 value exceeding zero are expected to exhibit heightened levels of plasma protein binding. Vdss is a crucial pharmacokinetic parameter of a drug, as it quantifies dispersion through various tissues within the body and influences the half-life and dosage interval. All designed compounds have a poor Vdss number and low BBB permeability, indicating a low possibility of central nervous system toxicity. Due to its contributions to drug disposition, transporter evaluations such as OATP1B1, OATP1B3, BCRP, and MRP1 are crucial. All designed compounds inhibit OATP1B1 and OATP1B3, but not BCRP. Compound A2 not MRP1 inhibitor while the rest of the compounds are [34].
Predicted metabolism parameters
The Cytochrome P450 (CYP450) enzyme superfamily is the primary enzyme group responsible for hepatic drug metabolism. CYP3A4 and CYP2D6 are the predominant isoforms, responsible for 75% of drug metabolism. Other isozymes, especially CYP1A2, CYP2C9, and CYP19, comprise the remainder. Considering the significant impact of metabolism on the drug's pharmacokinetic and drug-drug interaction profile, it is essential to ascertain the drug's metabolic profile. All the designed compounds are neither substrates nor inhibitors of these enzymes, indicating that the liver is not a primary route of clearance for these designed compounds. According to the ADMET results in table 4, all compounds are neither substrates nor inhibitors to CYP1A2, CYP2C19, CYP2D6, and CYP3A4. In the case of the CYP2C9 enzyme, compounds A2, A5, and K2 are classified as inhibitors. Additionally, to forecast stability against human liver microsomal enzymes (HLM), which are typically employed to evaluate drug clearance via the liver and to determine if a compound acts as a substrate or inhibitor of CYP isozymes, stable compounds must have an HLM value above 0.5. The results collected from ADMETlab 3.0 (table 4) indicate nine highly stable compounds, eight moderately stable compounds, and two unstable compounds (A4, A8). Notably, these two compounds have HLM values below the acceptable range, this may be a result of the R group, which is Cl in A4 and NH2 in A8 [35].
Table 4: Predicted metabolic profile of the designed compounds
Compound name |
HLM | CYP1A2 | CYP2C19 | CYP2C9 | CYP2D6 | CYP3A4 | |||||
| S | I | S | I | S | I | S | I | S | I | ||
| G | 0.407 | No | No | No | No | No | No | No | No | No | No |
| A1 | 0.539 | No | No | No | No | No | No | No | No | No | No |
| A2 | 0.861 | No | No | No | No | No | Yes | No | No | No | No |
| A3 | 0.569 | No | No | No | No | No | No | No | No | No | No |
| A4 | 0.092 | No | No | No | No | No | No | No | No | No | No |
| A5 | 0.436 | No | No | No | No | No | Yes | No | No | No | No |
| A6 | 0.477 | No | No | No | No | No | No | No | No | No | No |
| A7 | 0.474 | No | No | No | No | No | No | No | No | No | No |
| A8 | 0.196 | No | No | No | No | No | No | No | No | No | No |
| A9 | 0.781 | No | No | No | No | No | No | No | No | No | No |
| K1 | 0.889 | No | No | No | No | No | No | No | No | No | No |
| K2 | 0.951 | No | No | No | No | No | Yes | No | No | No | No |
| K3 | 0.796 | No | No | No | No | No | No | No | No | No | No |
| K4 | 0.719 | No | No | No | No | No | No | No | No | No | No |
| K5 | 0.481 | No | No | No | No | No | No | No | No | No | No |
| K6 | 0.57 | No | No | No | No | No | No | No | No | No | No |
| K7 | 0.94 | No | No | No | No | No | No | No | No | No | No |
| K8 | 0.729 | No | No | No | No | No | No | No | No | No | No |
| K9 | 0.989 | No | No | No | No | No | No | No | No | No | No |
Human Liver Microsomal enzymes; HLM, Cytochrome P450, CYP.
Predicted excretion and toxicity parameters
Plasma clearance (Clplasma) quantifies the body's ability to eliminate a drug, either by metabolism or renal excretion, by relating the drug elimination rate to the matching plasma concentration level. It is a vital parameter of pharmaceuticals, since it dictates the necessary dosage to sustain a stable plasma concentration. The predicted Clplasma of the designed compounds is less than 5 ml/min/kg, indicating a low Clplasma. The predicted results indicate that the main route of clearance is renal excretion. Another excretion parameter is the duration necessary for the plasma concentration to reduce to half its initial value (T1/2). It is a mixed concept influencing both clearance and volume of distribution (Vd). Designed compounds were anticipated to possess a T1/2 of less than 1.4 h. Concerning toxicity, none of the drugs were anticipated to inhibit the human ether-a-go-go-related gene (KCNH2), at IC50<10 μM (hERG 10). All compounds have results below 0.3, which means a low risk of inhibition (less than 0.3 low risk, more than 0.7 high risk). This gene encodes a voltage-gated potassium channel that plays a crucial role in regulating cardiac activity and resting potential exchange. Inhibiting this channel may result in long QT syndrome, arrhythmia, and Torsade de Pointes. Likewise, none of the target chemicals is anticipated to induce acute oral toxicity in rats. Generally, all designed compounds have an acceptable toxicity profile as shown in table 5 [36].
Molecular docking
The goal of molecular docking is to find the optimal orientation of ligands and their binding with the active site. The London dG (s-score), root mean square deviation (RMSD), indicating an average distance between the atoms of the posed ligand and the ligand for the investigated anti-cancer site, and generalized-born volume integral/weighted surface area free energy (GBVI/WSA dG) were utilized to assess the inhibitory effects of the designed compounds. These scores are the most common parameters used to determine the potential affinity and good fitting in the enzyme binding site, although these parameters give only a prediction that must be further investigated. These data are listed in table 6 [37].
The Molecular Operating Environment docking results assessed the efficacy of the compounds as carbonic anhydrase XII and IX enzyme inhibitors; the high s-score and GBVI/WSA dG numbers revealed that the examined compound has a favorable binding affinity with the target protein.
Carbonic anhydrase XII results
The reference compound acetazolamide (AZM) shows an s-score of-6.0260, GBVI/WSA dG-8.6086, and an RMSD of 1.9818 with only two hydrogen bonds with the active site, as shown in fig. 4, while all the designed compounds show higher s-score than acetazolamide and acceptable RMSD and GBVI/WSA dG values, as table 6 shows. Compound G shows-7.5142 s-score,-9.6178 GBVI/WSA dG, 1.8375 RMSD, and has four bindings with the hCA site (Zn901, Thr199, Thr199, and Thr200), and this is the highest result of this study. The rest of the results are listed in table 6.
Table 5: Predicted excretion and toxicity of the designed compounds
| Compound name | Clplasma | T1/2 | hERG 10 | Carcino-genicity | H-HT | AMES mutagenicity | Rat oral acute toxicity |
| G | 1.646 | 1.206 | 0.133 | 0.759 | 0.988 | 0.519 | 0.09 |
| A1 | 1.951 | 1.099 | 0.146 | 0.756 | 0.988 | 0.482 | 0.08 |
| A2 | 2.507 | 1.09 | 0.139 | 0.827 | 0.981 | 0.613 | 0.128 |
| A3 | 1.702 | 1.327 | 0.141 | 0.775 | 0.986 | 0.496 | 0.102 |
| A4 | 1.02 | 1.357 | 0.215 | 0.7 | 0.988 | 0.346 | 0.102 |
| A5 | 0.936 | 1.391 | 0.171 | 0.746 | 0.984 | 0.288 | 0.132 |
| A6 | 1.083 | 1.304 | 0.154 | 0.854 | 0.983 | 0.612 | 0.28 |
| A7 | 1.569 | 1.229 | 0.17 | 0.884 | 0.987 | 0.936 | 0.276 |
| A8 | 1.557 | 1.357 | 0.112 | 0.939 | 0.966 | 0.914 | 0.264 |
| A9 | 2.97 | 1.047 | 0.088 | 0.952 | 0.976 | 0.795 | 0.25 |
| K1 | 2.405 | 1.083 | 0.122 | 0.771 | 0.989 | 0.519 | 0.089 |
| K2 | 2.59 | 1.039 | 0.135 | 0.839 | 0.985 | 0.608 | 0.142 |
| K3 | 1.818 | 1.181 | 0.129 | 0.756 | 0.984 | 0.536 | 0.122 |
| K4 | 1.259 | 1.348 | 0.176 | 0.745 | 0.988 | 0.414 | 0.102 |
| K5 | 1.276 | 1.26 | 0.139 | 0.769 | 0.98 | 0.391 | 0.155 |
| K6 | 1.37 | 1.301 | 0.145 | 0.754 | 0.989 | 0.587 | 0.238 |
| K7 | 1.937 | 1.164 | 0.146 | 0.852 | 0.984 | 0.885 | 0.282 |
| K8 | 1.831 | 1.289 | 0.118 | 0.889 | 0.977 | 0.831 | 0.203 |
| K9 | 2.949 | 1.108 | 0.096 | 0.9 | 0.981 | 0.663 | 0.254 |
Plasma clearance, Clplasma; T1/2, Half-life, human ether-a-go-go-related gene at IC50<10μM (hERG 10); human Hepatotoxic, H-HT.
Table 6: Molecular docking results using hCA XII (using MOE 2015.1)
| Compound name | R group and its position | S-score | dG | RMSD | No. of bonds | Binding amino acids |
| AZM | ------ | -6.0260 | -8.6086 | 1.9818 | 2 | Zn901 and Thr199 |
| SLC-0111 | ------ | -7.1162 | -9.7571 | 1.6759 | 3 | Zn901, Thr200, and Asn62 |
| G | H | -7.5142 | -9.6178 | 1.8375 | 4 | Zn901, Thr199, Thr199, and Thre200 |
| A1 | Para-CH3 | -7.1281 | -9.5236 | 1.7679 | 3 | Zn901, Thr199 and His94 |
| A2 | Para-OCH3 | -7.4132 | -9.5700 | 1.8877 | 5 | Zn901, Thr199, Thr200, His94 and Pro201 |
| A3 | Para-OH | -7.1219 | -9.3308 | 1.9155 | 5 | Zn901, Thr199, Thr200, His94 and Pro201 |
| A4 | Para-Cl | -7.2370 | -9.1454 | 1.6793 | 4 | Zn901, Thr199, Thr200, and His94 |
| A5 | Para-Br | -7.1522 | -8.7834 | 1.6894 | 4 | Zn901, Thr199, Thr200, and Pro201 |
| A6 | Para-F | -7.3601 | -9.4584 | 1.4926 | 4 | Zn901, Thr199, Thr199, and Thre200 |
| A7 | Para-NO2 | -7.4331 | -9.6084 | 1.7289 | 4 | Zn901, Thr199, Thr200, and His94 |
| A8 | Para-NH2 | -7.7812 | -9.6548 | 1.8519 | 5 | Zn901, Thr199, Thr200, His94 and Pro201 |
| A9 | Para-N(CH3)2 | -7.2811 | -8.7500 | 1.9410 | 4 | Zn901, Thr199, Thr200, and Pro201 |
| K1 | Meta-CH3 | -6.4287 | -8.6017 | 1.3222 | 1 | Zn901 |
| K2 | Meta-OCH3 | -6.9408 | -8.5684 | 1.8331 | 4 | Zn901, Thr199, Thr199, and Thre200 |
| K3 | Meta-OH | -7.0768 | -9.7421 | 1.5081 | 2 | Zn901 and Thr199 |
| K4 | Meta-F | -6.7477 | -8.9393 | 1.4493 | 3 | Zn901, Thr199 and His94 |
| K5 | Meta-Br | -7.0381 | -9.0328 | 1.7443 | 2 | Zn901 and Thr199 |
| K6 | Meta-Cl | -7.4959 | -9.2290 | 1.2692 | 4 | Zn901, Thr199, Thr200, and Gln94 |
| K7 | Meta-NO2 | -7.2513 | -9.3945 | 1.5570 | 3 | Zn901, Thr199 and Asn62 |
| K8 | Meta-NH2 | -6.8263 | -9.0788 | 1.2692 | 2 | Zn901 and Thr199 |
| K9 | Meta-N(CH3)2 | -7.1558 | -9.3181 | 1.6021 | 3 | Zn901, Thr199 and Lys67 |
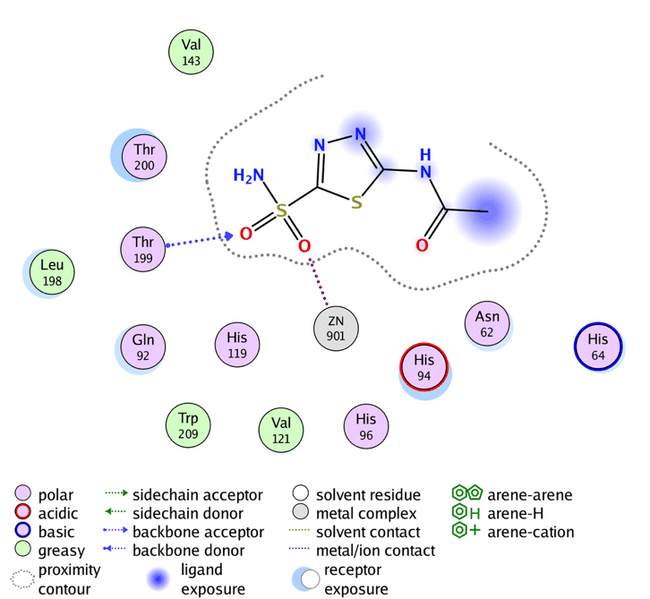
Fig. 4: Acetazolamide binding with human carbonic anhydrase XII (PDB code: 1JD0)
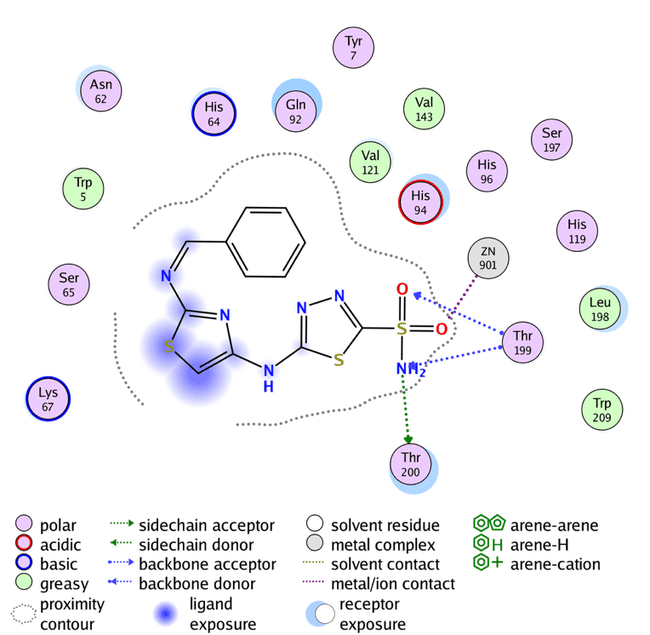
Fig. 5: Compound G binding with human carbonic anhydrase XII (PDB code: 1JD0)
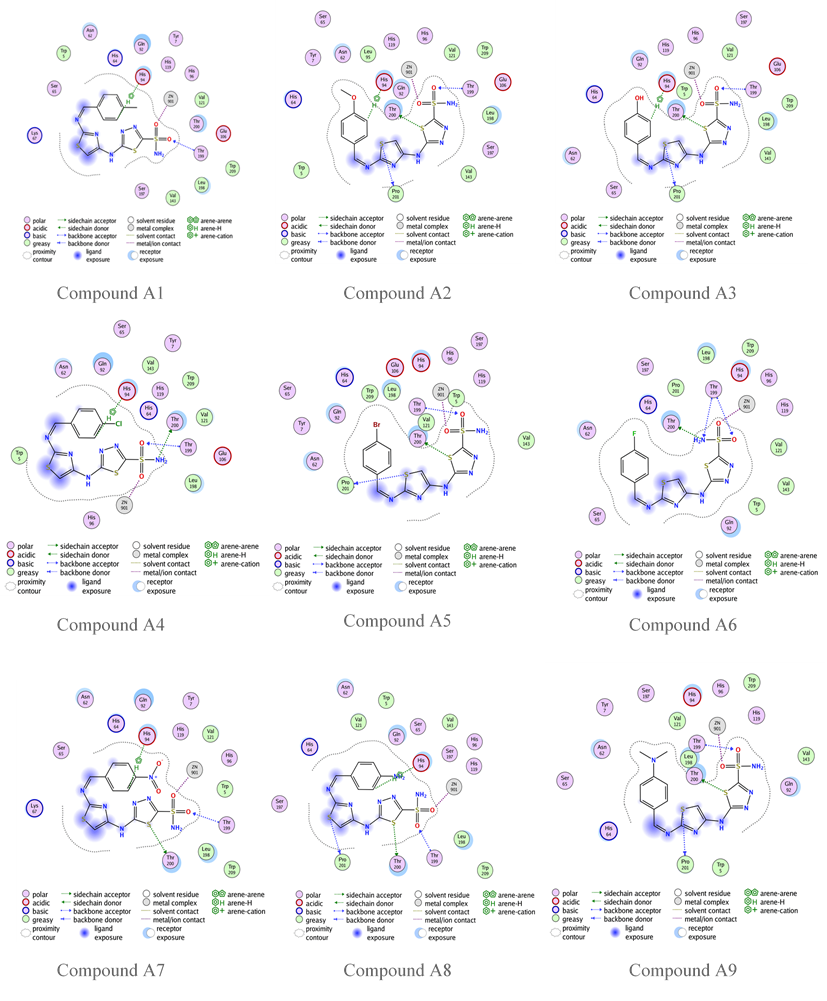
Fig. 6: Compounds A1-A9 binding with human carbonic anhydrase XII (PDB code: 1JD0)
The previous molecular docking results listed in table 6 indicate that the highest s-score occurs when the designed compound has no substitutions on the aromatic ring of benzaldehyde, seen in compound G, and the highest GBVI/WSA dG result occurs in compound K3 (with Meta-OH group). Compounds A1, A2, A7, and A8 also show promising GBVI/WSA dG values, above-9.5 which is very close to the positive control SLC-0111. However, the docking results of all designed compounds show a good binding affinity, and more favorable orientations comparing to the reference compound acetazolamide, fig. 4 shows that acetazolamide bind to the active site by only two hydrogen bonds, this demonstrates that thiazole ring that incorporated to our designed compounds plays a significant role in binding affinity with the enzyme, providing flexibility and enhancing the likelihood of interaction with the enzyme's active site. All the designed compounds possess fundamental interactions, particularly the zinc-sulfonamide bond, which is vital in maintaining the inhibitory efficacy of these compounds. Hydrogen bindings with threonine 200 and 199 are important too, but not essential, and many of the designed compounds show this type of bonding, as shown in fig. 6 and 7 [38]. Additionally, the influence of position and nature of benzaldehyde substitution have a significant effect on the binding orientation, for example compound A8 has s-score and GBVI/WSA dG results higher than the meta-substituted analogs this suggest that-NH2 group in para position give that compound the perfect fit for the enzyme’s active site, but when the same group is in the meta position, s-score and GBVI/WSA dG values decrease significantly, indicating that compound K8, has poor fit with the active site. Otherwise, groups like-Cl and-N(CH3)2 have a good fit in both para and meta positions, as shown in table 6 with compounds A6 and K6, respectively, demonstrating that these groups are important in the binding between the compounds and the enzyme’s active site. RMSD low values of compound K6 and K8 hint a possible overfitting of docking poses with the reference compound.
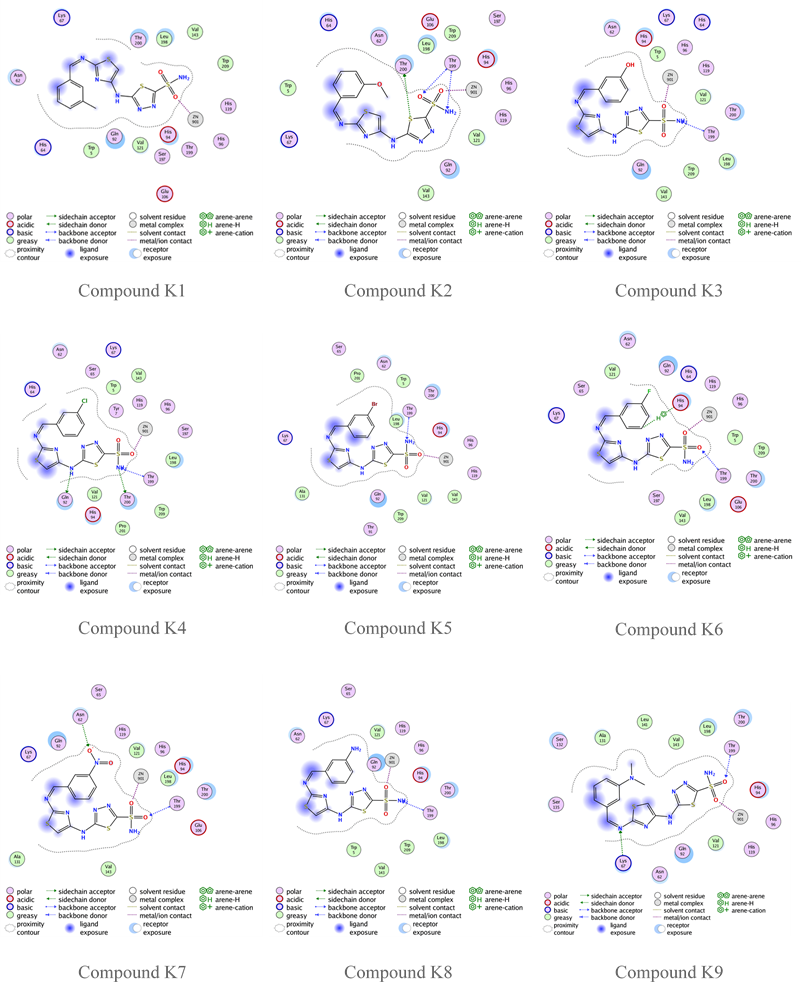
Fig. 7: Compounds K1-K9 binding with human carbonic anhydrase XII (PDB code: 1JD0)
Carbonic anhydrase IX results
The reference compound acetazolamide (AZM) shows an s-score of-6.0888, GBVI/WSA dG-8.5072, and an RMSD of 0.9290 with only two hydrogen bonds with the active site, as shown in fig. 8. In contrast, all the designed compounds exhibit a higher s-score than acetazolamide, with acceptable RMSD and GBVI/WSA dG values, as shown in table 7. Compound A2 shows-7.7122 s-score, which is the highest value. While the highest GBVI/WSA dG value occurs with compound A5, these compounds, A2 and A5, have an R group of OCH3 and Br, respectively, highlighting the importance of the nature and location of the R group. The remaining results are listed in table 7.
Compounds A2, A4, A6, A7, A9, K1, K2, K5, K7, K8, and K9 have an s-score close to the positive reference compound SLC-0111, indicating a good fit in the pocket of hCA IX enzyme. On the other hand, the GBVI/WSA dG results show that compounds G, A3, A4, A5, A6, A7, A9, K3, K4, K6, K7, and K8 have excellent results. Compound K5 and K9 have the lowest docking results,-8.5687 and-8.3376, respectively. Compound G 2D binding with the active site is highlighted in fig. 9, while fig. 10 and 11 highlight the 2D binding with the active site of the rest of the designed compounds.
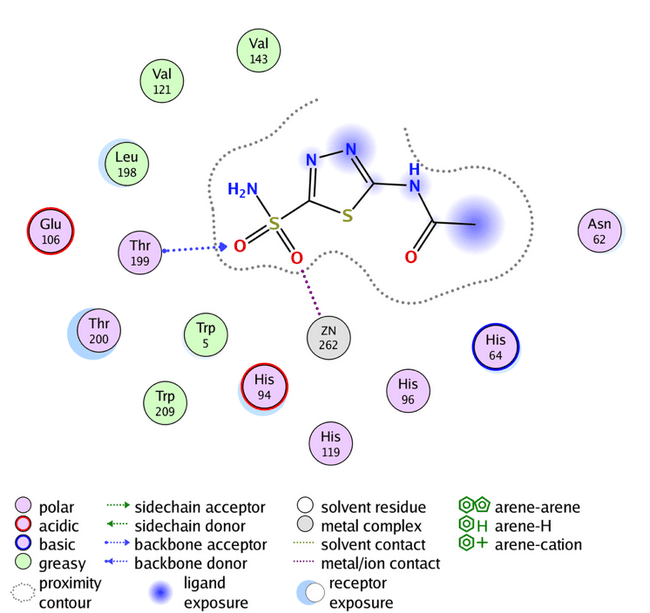
Fig. 8: Acetazolamide binding with human carbonic anhydrase IX (PDB code: 3IAI)
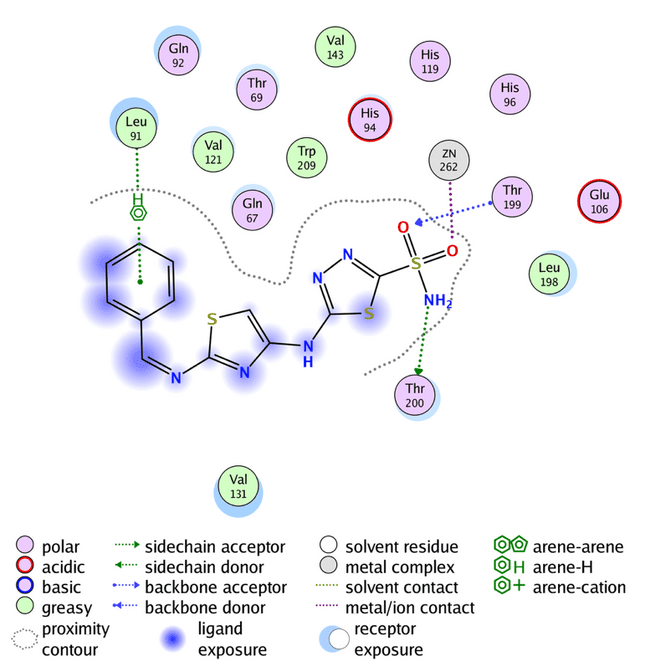
Fig. 9: Compound G binding with human carbonic anhydrase IX (PDB code: 3IAI)
Table 7: Molecular docking results using hCA IX (using MOE 2015.1)
| Compound name | R group and it's position | S-score | dG | RMSD | No. of bonds | Binding amino acids |
| AZM | ------ | -6.0888 | -8.5072 | 0.9290 | 2 | ZN262 and Thr199 |
| SLC-0111 | ------ | -7.8401 | -9.4345 | 1.5462 | 5 | ZN262, Thr199, Thr200, Gln92, and Gln92 |
| G | H | -6.7858 | -9.2705 | 1.2969 | 4 | ZN262, Thr199, Thr200, and Leu91 |
| A1 | Para-CH3 | -6.9973 | -8.9321 | 1.4183 | 4 | ZN262, Thr199, Thr200, and Leu91 |
| A2 | Para-OCH3 | -7.7122 | -8.8309 | 1.5822 | 3 | ZN262, His94, and His64 |
| A3 | Para-OH | -6.8993 | -9.3194 | 1.5631 | 2 | ZN262 and His94 |
| A4 | Para-Cl | -7.1388 | -9.1300 | 1.4074 | 3 | ZN262, Thr199, and Leu198 |
| A5 | Para-Br | -6.9830 | -9.7895 | 1.5437 | 3 | ZN262, Thr199, and Thr200 |
| A6 | Para-F | -7.2798 | -9.0632 | 0.6833 | 4 | ZN262, His64, Pro201, and Trp5 |
| A7 | Para-NO2 | -7.5308 | -9.0672 | 1.0796 | 5 | ZN262, Thr199, Leu198, Asn62, and Trp5 |
| A8 | Para-NH2 | -6.7647 | -8.9707 | 1.7729 | 1 | ZN262 |
| A9 | Para-N(CH3)2 | -7.6829 | -9.7380 | 1.8818 | 3 | ZN262, Thr199, and Trp5 |
| K1 | Meta-CH3 | -7.0160 | -8.9181 | 1.7602 | 3 | ZN262, His94, and Gln67 |
| K2 | Meta-OCH3 | -7.4418 | -8.7105 | 1.7663 | 3 | ZN262, Thr199, and His64 |
| K3 | Meta-OH | -6.8269 | -9.4528 | 1.4219 | 3 | ZN262, Thr200, and Val121 |
| K4 | Meta-F | -6.9146 | -9.6569 | 1.7089 | 6 | ZN262, Thr199, Thr199, Thr200, His94, and Leu198 |
| K5 | Meta-Br | -7.1686 | -8.5687 | 1.8664 | 4 | ZN262, His94, Gln92, and Gln92 |
| K6 | Meta-Cl | -6.9967 | -9.1392 | 1.5259 | 4 | ZN262, Thr199, Asn62, and His64 |
| K7 | Meta-NO2 | -7.2413 | -9.7468 | 1.5479 | 3 | ZN262, Thr199, and Asn62 |
| K8 | Meta-NH2 | -7.0996 | -9.2460 | 1.3377 | 3 | ZN262, Thr199, and Arg60 |
| K9 | Meta-N(CH3)2 | -7.5427 | -8.3376 | 1.6772 | 6 | ZN262, Thr199, His94, His64, His64 and Arg60 |
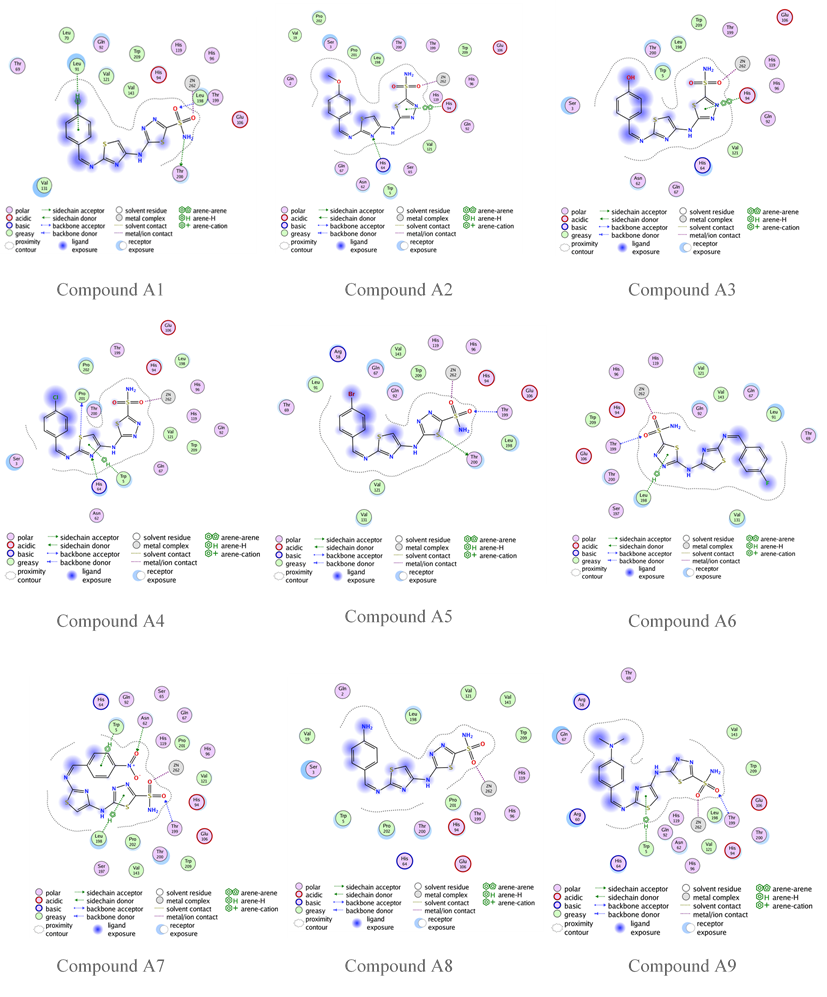
Fig. 10: Compounds A1-A9 binding with human carbonic anhydrase IX (PDB code: 3IAI)
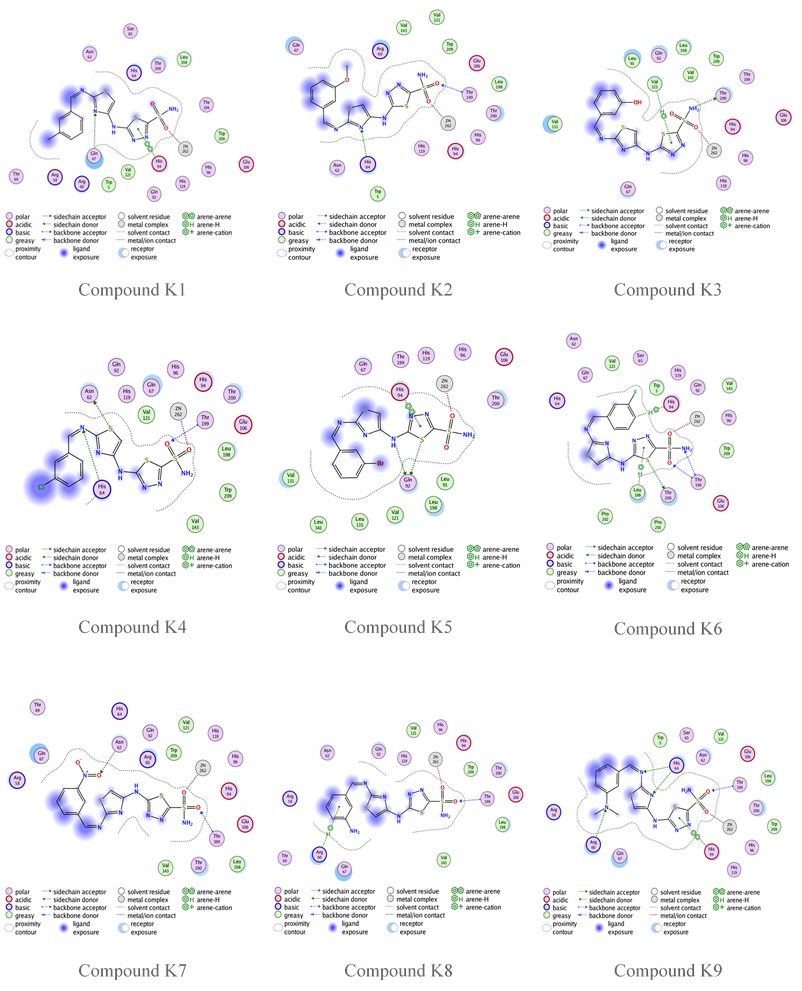
Fig. 11: Compounds K1-K9 binding with human carbonic anhydrase IX (PDB code: 3IAI)
CONCLUSION
This research focuses on the development of novel acetazolamide analogues bearing a thiazole moiety, followed by an assessment of their potential as carbonic anhydrase inhibitors for the treatment of breast cancer by molecular docking methods. The Molecular Operating Environment docking analysis indicated that these designed analogues have this potential.
ACKNOWLEDGMENT
The authors express their profound gratitude to the faculty and the employees of the University of Kufa's pharmaceutical chemistry department.
AUTHORS CONTRIBUTIONS
All authors have contributed equally
CONFLICT OF INTERESTS
Declared none
REFERENCES
Brown JS, Amend SR, Austin RH, Gatenby RA, Hammarlund EU, Pienta KJ. Updating the definition of cancer. Mol Cancer Res. 2023 Nov 1;21(11):1142-7. doi: 10.1158/1541-7786.MCR-23-0411, PMID 37409952.
Castaneda M, Den Hollander P, Kuburich NA, Rosen JM, Mani SA. Mechanisms of cancer metastasis. Semin Cancer Biol. 2022 Dec;87:17-31. doi: 10.1016/j.semcancer.2022.10.006, PMID 36354098.
Ghosh T, Maity TK, Singh J. Evaluation of antitumor activity of stigmasterol a constituent isolated from Bacopa monnieri linn aerial parts, against Ehrlich ascites carcinoma in mice. Orient Pharm Exp Med. 2011 Mar;11(1):41-9. doi: 10.1007/s13596-011-0001-y.
National Center for Health Statistics. National Vital Statistics Reports Hyattsville MD: National Center for Health Statistics; 2021 Jul. Available from: https://www.cdc.gov/nchs/ data/nvsr/nvsr70/nvsr70_09-508.pdf. [Last accessed on 26 Nov 2025].
Madriwala B, BVS, Jays J. Molecular docking study of hentriacontane for anticancer and antitubercular activity. Int J Chem Res. 2022 Oct 1;6(4):1-4. doi: 10.22159/ijcr.2022v6i4.208.
Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024 May;74(3):229-63. doi: 10.3322/caac.21834, PMID 38572751.
Brennan P, Davey Smith G. Identifying novel causes of cancers to enhance cancer prevention: new strategies are needed. J Natl Cancer Inst. 2022 Mar 8;114(3):353-60. doi: 10.1093/jnci/djab204, PMID 34743211.
Zhu W, Xie L, Han J, Guo X. The application of deep learning in cancer prognosis prediction. Cancers. 2020 Mar 5;12(3):603. doi: 10.3390/cancers12030603, PMID 32150991.
Liao C, Liu X, Zhang C, Zhang Q. Tumor hypoxia: from basic knowledge to therapeutic implications. Semin Cancer Biol. 2023 Jan;88:172-86. doi: 10.1016/j.semcancer.2022.12.011, PMID 36603793.
Weiss F, Lauffenburger D, Friedl P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat Rev Cancer. 2022 Mar;22(3):157-73. doi: 10.1038/s41568-021-00427-0, PMID 35013601.
Su Z, Dong S, Zhao SC, Liu K, Tan Y, Jiang X. Novel nanomedicines to overcome cancer multidrug resistance. Drug Resist Updat. 2021 Sep;58:100777. doi: 10.1016/j.drup.2021.100777, PMID 34481195.
Campestre C, De Luca V, Carradori S, Grande R, Carginale V, Scaloni A. Carbonic anhydrases: new perspectives on protein functional role and inhibition in Helicobacter pylori. Front Microbiol. 2021 Mar 19;12:629163. doi: 10.3389/fmicb.2021.629163, PMID 33815311.
Pandey V. Modeling of carbonic anhydrase (II). Int J Pharm Pharm Sci. 2018 Jan 1;10(1):202. doi: 10.22159/ijpps.2018v10i1.22775.
Kciuk M, Gielecinska A, Mujwar S, Mojzych M, Marciniak B, Drozda R. Targeting carbonic anhydrase IX and XII isoforms with small molecule inhibitors and monoclonal antibodies. J Enzyme Inhib Med Chem. 2022 Dec 31;37(1):1278-98. doi: 10.1080/14756366.2022.2052868, PMID 35506234.
Li Y, Lei B, Zou J, Wang W, Chen A, Zhang J. High expression of carbonic anhydrase 12 (CA12) is associated with good prognosis in breast cancer. Neoplasma. 2019;66(3):420-6. doi: 10.4149/neo_2018_180819N624, PMID 30784287.
Shin HJ, Rho SB, Jung DC, Han IO, Oh ES, Kim JY. Carbonic anhydrase IX (CA9) modulates tumor-associated cell migration and invasion. J Cell Sci. 2011 Apr 1;124(7):1077-87. doi: 10.1242/jcs.072207, PMID 21363891.
Cuffaro D, Nuti E, Rossello A. An overview of carbohydrate-based carbonic anhydrase inhibitors. J Enzyme Inhib Med Chem. 2020 Jan 1;35(1):1906-22. doi: 10.1080/14756366.2020.1825409, PMID 33078634.
Ovung A, Bhattacharyya J. Sulfonamide drugs: structure antibacterial property toxicity and biophysical interactions. Biophys Rev. 2021 Apr;13(2):259-72. doi: 10.1007/s12551-021-00795-9, PMID 33936318.
Shukralla AA, Dolan E, Delanty N. Acetazolamide: old drug, new evidence? Epilepsia Open. 2022 Sep;7(3):378-92. doi: 10.1002/epi4.12619, PMID 35673961.
Chhabria MT, Patel S, Modi P, Brahmkshatriya PS. Thiazole: a review on chemistry synthesis and therapeutic importance of its derivatives. Curr Top Med Chem. 2016 Sep 2;16(26):2841-62. doi: 10.2174/1568026616666160506130731, PMID 27150376.
De Santana TI, Barbosa MO, Gomes PA, Da Cruz AC, Da Silva TG, Leite AC. Synthesis anticancer activity and mechanism of action of new thiazole derivatives. Eur J Med Chem. 2018 Jan;144:874-86. doi: 10.1016/j.ejmech.2017.12.040, PMID 29329071.
Dong J, Wang NN, Yao ZJ, Zhang L, Cheng Y, Ouyang D. ADMET lab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J Cheminform. 2018 Dec;10(1):29. doi: 10.1186/s13321-018-0283-x, PMID 29943074.
Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021 Jul 2;49(W1):W5-14. doi: 10.1093/nar/gkab255, PMID 33893803.
Al Gburi K, Naser NH, Jasmi M. Design and computational analysis of new isatin-imine hybrids as selective HDAC6 inhibitors. Int J App Pharm. 2025 May 7;17(3):214-27. doi: 10.22159/ijap.2025v17i3.53658.
Wei Y, Palazzolo L, Ben Mariem O, Bianchi D, Laurenzi T, Guerrini U. Investigation of in silico studies for cytochrome P450 isoforms specificity. Comput Struct Biotechnol J. 2024 Dec;23:3090-103. doi: 10.1016/j.csbj.2024.08.002, PMID 39188968.
Shah R, Alharbi A, Hameed AM, Saad F, Zaky R, Khedr AM. Synthesis and structural elucidation for new Schiff base complexes; conductance conformational MOE-docking and biological studies. J Inorg Organomet Polym. 2020 Sep;30(9):3595-607. doi: 10.1007/s10904-020-01505-w.
Aloufi BH. Structure-based multi-targeted molecular docking and molecular dynamic simulation analysis to identify potential inhibitors against ovarian cancer. J Biochem Technol. 2022;13(2):29-39. doi: 10.51847/b1KFmETha6.
Alibeg AA, Hussein TS. In silico study of new isatin sulfonamide derivatives as carbonic anhydrase inhibitors. Wiad Lek. 2024 Oct 30;77(10):2027-32. doi: 10.36740/WLek/193997, PMID 39661898.
Ononamadu CJ, Abdalla M, Ihegboro GO, Li J, Owolarafe TA, John TD. In silico identification and study of potential anti-mosquito juvenile hormone binding protein (MJHBP) compounds as candidates for dengue virus vector insecticides. Biochem Biophys Rep. 2021 Dec;28:101178. doi: 10.1016/j.bbrep.2021.101178, PMID 34901473.
Whittington DA, Waheed A, Ulmasov B, Shah GN, Grubb JH, Sly WS. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc Natl Acad Sci USA. 2001 Aug 14;98(17):9545-50. doi: 10.1073/pnas.161301298, PMID 11493685.
Arsianti AA, Astuty H, Fadilah F, Bahtiar A, Tanimoto H, Kakiuchi K. Design and screening of gallic acid derivatives as inhibitors of malarial dihydrofolate reductase by in silico docking. Asian J Pharm Clin Res. 2017 Feb 1;10(2):330. doi: 10.22159/ajpcr.2017.v10i2.15712.
Ward SE, Davis A, editors. The Handbook of Medicinal Chemistry: Principles and Practice. 2nd ed. Cambridge: Royal Society of Chemistry; 2023. Available from: https://books.rsc.org/books/monograph/2061.
Williams J, Siramshetty V, Nguyen DT, Padilha EC, Kabir M, Yu KR. Using in vitro ADME data for lead compound selection: an emphasis on Pampa pH 5 permeability and oral bioavailability. Bioorg Med Chem. 2022 Feb;56:116588. doi: 10.1016/j.bmc.2021.116588, PMID 35030421.
Klimoszek D, Jelen M, Dolowy M, Morak Mlodawska B. Study of the lipophilicity and ADMET parameters of new anticancer diquinothiazines with pharmacophore substituents. Pharmaceuticals (Basel). 2024 Jun 3;17(6):725. doi: 10.3390/ph17060725, PMID 38931392.
Sohlenius Sternbeck AK, Terelius Y. Evaluation of ADMET predictor in early discovery drug metabolism and pharmacokinetics project work. Drug Metab Dispos. 2022 Feb;50(2):95-104. doi: 10.1124/dmd.121.000552, PMID 34750195.
Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021 Jul 2;49(W1):W5-14. doi: 10.1093/nar/gkab255, PMID 33893803.
Konyar D, Okur H, Arslan Z. Molecular docking studies of cox inhibitors on wild-type ras. Ank Univ Eczacılık Fak Derg. 2021 Oct 22;46(1):23-34. doi: 10.33483/jfpau.1008048.
Barua H, Bhagat N, Toraskar MP. Study of binding interactions of human carbonic anhydrase XII. Int J Curr Pharm Sci. 2016 Dec 31;9(1):118. doi: 10.22159/ijcpr.2017v9i1.16633.