
Int J App Pharm, Vol 18, Issue 2, 2026, 326-337Original Article
INNOVATIVE DISEASE MODIFYING TREATMENTS: EVALUATING THE ATRIGEL® SYSTEM FOR ENHANCED THERAPEUTIC EFFICIENCY
SUSHANT ISHWAR POTE1 OM SAMBHAJI SHELKE2*, RAJESH KHATHURIYA1, VRUSHALI A. KULKARNI4, SACHIN J. FARTADE5
1Department of Pharmaceutics, Pacific Academy of Higher Education and Research University, Udaipur, Rajasthan, India. 2Department of Pharmaceutics, Manipur International University, Imphal, Manipur-795140, India. 3Sinomune Pharmaceutical Co., Ltd, Wuxi, Jiangsu-214194, China. 4Department of Pharmaceutics, Amepurva Forum’s Nirant Institute of Pharmacy, Boramani, Solapur, Maharashtra, India. 5Department of Pharmaceutics, Dr VVPF’s College of Pharmacy, Viladghat, Ahilyanagar, Maharashtra, India
*Corresponding author: Om Sambhaji Shelke; *Email: om.shelke20@gmail.com
Received: 19 Jul 2025, Revised and Accepted: 27 Dec 2025
ABSTRACT
Methods: A 3² randomised complete factorial design was employed to optimise critical formulation parameters such as drug-to-polymer ratio and solvent concentration. The Atrigel® system was formulated using N-methyl pyrrolidone (NMP) and PLGA. The developed formulations were characterised for syringeability, viscosity, and in vitro drug release. Physicochemical properties were assessed, and biological evaluations included ex vivo release studies and cytotoxicity assays using the hen drumstick model and L929 fibroblasts, respectively.
Results: The optimised formulation achieved sustained drug release with reduced dosing frequency. Statistical analysis confirmed significant effects of drug-to-polymer ratio and solvent concentration on release kinetics (p<0.05). An amorphous GA dispersion was formed within PLGA, which exhibited good physical compatibility. The Higuchi model's high slope (5.3858) and negative intercept (-4.8536) corroborate the strong linear relationship between drug release and the square root of time, providing compelling evidence for classically diffusion-controlled release. Ex vivo studies demonstrated consistent release profiles (p>0.05 vs. in vitro), and cytotoxicity assays showed high biocompatibility.
Conclusion: The Atrigel® system offers a promising long-acting alternative to conventional GA therapy, with controlled release, enhanced stability, and improved patient compliance. Future studies should focus on in vivo pharmacokinetics and scalability.
Keywords: Atrigel, Depot injection, Glatiramer acetate, Multiple sclerosis, Sustained-release, PLGA
© 2026 The Authors.Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2026v18i2.56159 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Multiple sclerosis (MS) is an immunopathological, non-traumatic, disabling, chronic autoimmune inflammatory neurological disease that is influenced by genetic and environmental factors and is one of the most common causes of neurological disability in young adults [1–3]. The prevalence of MS has increased in both developed and developing countries. More than twice as many women are affected as men [4, 5]. MS primarily affecting young adults falls into four main clinical types: Relapsing-remitting MS (RRMS), Secondary-progressive MS (SPMS), Primary-progressive MS (PPMS), and Progressive-relapsing MS (PRMS). Around 85% of MS patients initially present with RRMS. This type of MS is marked by acute neurological episodes that occur intermittently, with symptoms that improve over days to weeks, either entirely or partially [6, 7]. In most patients, the MS is characterised initially by episodes of reversible neurological deficits. The axonal damage was recognised as an early event in the disease process and as an essential determinant of long-term disability [8, 9]. Diagnosing MS involves identifying inflammatory and demyelinating damage within the central nervous system (CNS) that occurs across multiple areas and over time. Tests such as cerebrospinal fluid (CSF) analysis, evoked potential recording, urodynamic studies of bladder function, and ocular coherence tomography (OCT) can assist in diagnosing individual cases but are frequently not required [10-12]. Currently, several Disease-modifying treatments (DMTs) are available for the treatment of RRMS, and their primary purpose is to reduce the frequency of relapses and the severity of CNS inflammation [13]. There is no cure for MS to date. New, more effective treatments for MS therapy are now available. After years of relying on DMTs such as interferon beta (IFNβ) and GA as primary options, fingolimod emerged as the first oral DMT, receiving approval in the United States in 2010 [14-16] The beneficial effect of GA is not only limited to its impact on neurons but also involves a reduction of demyelination and an increase of myelin repair [17].
GA is a complex, heterogeneous mixture of polypeptides with immunomodulatory activity, approved in 1996 for the treatment of relapsing multiple sclerosis in the United States under the brand name COPAXONE, and in 2001 in Europe [18, 19]. GA is a complex mixture of polypeptides composed of four amino acids resembling Myelin Basic Protein (MBP). It is produced by polymerising the amino acids L-glutamic acid, L-alanine, L-lysine, and L-tyrosine, followed by partial hydrolysis [20, 21].
Atrigel utilises a biodegradable polymer solution that, upon injection, forms a solid or semi-solid depot upon contact with bodily fluids, enabling controlled and sustained release of therapeutic agents. Atrigel offers significant advantages, including reduced dosing frequency, improved patient compliance, enhanced drug stability, and localised delivery, minimising systemic side effects. Its applications in current injection systems span a wide range of medical fields, such as pain management (e. g., extended-release bupivacaine), hormone therapy (e. g., leuprolide acetate for prostate cancer), and post-surgical recovery, where prolonged drug release is critical. The technology’s ability to deliver both small molecules and biologics with precision makes it a superior alternative to traditional bolus injections, which often result in rapid drug clearance and fluctuating plasma concentrations. Atrigel’s versatility and efficiency position it as a transformative solution in long-term therapeutic delivery [22, 23].
GA is indicated for the treatment of MS, including clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease. In adults, it is administered as a 20 mg/ml daily injection or a 40 mg/ml three times a week subcutaneous [22]. While daily injections of GA have been the standard, patient compliance remains a significant challenge. This study addresses these issues by proposing a long-acting Atrigel® formulation. MS necessitates lifelong therapy, which presents substantial challenges in terms of adherence. Despite the development and approval of various disease-modifying treatments, there is still a pressing need to enhance patient outcomes by improving treatment efficacy, tolerability, and adherence [23, 24].
In this study, a stable, efficacious GA long-acting in-situ forming implants called Atrigel containing polymer and solvent were evaluated for improving treatment efficacy, as represented in fig. 1.
MATERIALS AND METHODS
Materials
GA was obtained from Pharmaffiliates in Haryana, India. Resomer 202H (75:25) and other grades of PLGA polymers were obtained as a gift sample from Evonik India. Resomer 202H (75:25) was selected for its known biocompatibility and controlled degradation profile, making it ideal for sustained release formulations. NMP, Dimethyl Sulfoxide (DMSO), Ethyl Acetate (EA), and Dichloromethane (DCM) were purchased from Sigma Aldrich, and Mannitol was obtained from DFE Pharma. pH 7.4 Phosphate buffer saline was purchased from Merck India. Sodium azide was purchased from CDH Finechem. Multiple pH calibration solutions from Thermo Orion were used. Penicillin, streptomycin, fetal bovine sera (FBS), Roswell Park Memorial Institute (RPMI) 1640 culture medium, and trypsin were obtained from Gibco (Germany). Mouse L929 fibroblast cell line issued by the National Centre for Cell Science.
Preparation of atrigel
For the preparation of atrigel, an accurately weighed quantity of resomer 202H, 2.5 g, was added to a vial, and 7.5 g of solvent NMP were added to the same vial. The vial was sealed with a rubber stopper and a flip-of-seal. The polymer was allowed to dissolve in the solvent. Once the polymer was soaked completely, it was checked for clarity and hydration. Before administration, the polymer phase was mixed with GA powder. After complete mixing, the in situ forming implant was ready for administration [25, 26].
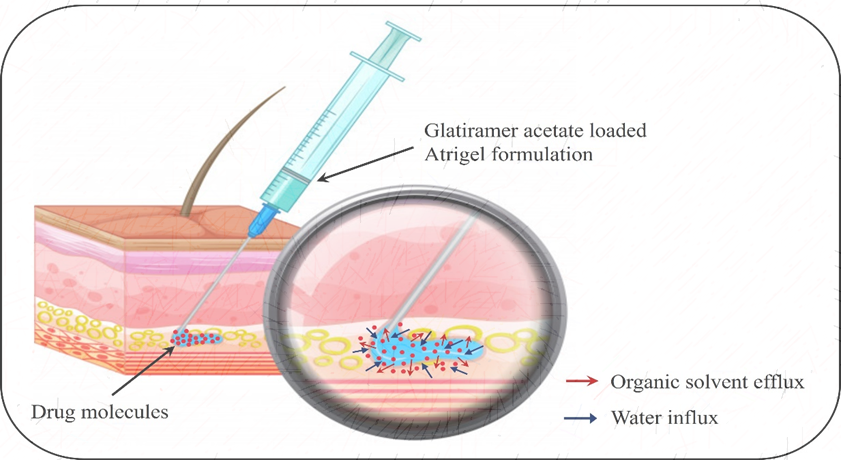
Fig. 1: Mechanism of in situ forming implant via atrigel system
Formulation optimization
In the initial experiments, a combination of PLGA polymers with both acid and ester end groups, such as PLGA 50:50, PLGA 75:25, and PLGA 85:15, was employed. The organic solvents NMP, DMSO, EA, and DCM were used in the formulations. Preliminary research studies were conducted to assess the influence of key formulation and process parameters, including drug-to-polymer ratio and solvent concentration. The critical response variables studied included syringeability, measured via break-loose force (BLF) and glide force (GF), viscosity, and drug release kinetics. To systematically optimise these factors, a 32 randomised complete factorial design (9 trials) and one centre point were employed, examining two independent variables, drug-to-polymer ratio (X₁) and solvent concentration (X₂), each at three levels (low, medium, high) within predefined experimental ranges. All dependent variables were thoroughly analysed to determine the effects of these formulation parameters on the system's performance [27, 28].
The independent variables and their respective levels are presented in table 1. Nine experimental runs, including one centre point, were prepared and systematically evaluated with respect to the critical quality attributes (CQAs) of GF (Y₁), viscosity (Y₂), and in vitro drug release (Y₃). Optimisation studies and subsequent data analysis, including the generation of response surface plots, were conducted using Design Expert® software (Version 13.0.5.0, State-Ease Inc., Minneapolis, MN).
Table 1: 32 Randomised full factorial design parameters and experimental conditions
| Independent variables | X1: Drug to polymer ratio (mg) | X2: Solvent concentration (mg) |
| Levels | Low (-1) | 20:20 |
| Medium (0) | 20:100 | |
| High (+1) | 20:200 | |
| Dependent variables | Y1: GF (N) Y2: Viscosity (cP) Y3: Release (%) |
The experimentally observed response values were compared with the expected values to confirm the experimental design. The relative error (%) was determined using the following equation:
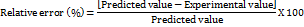
Characterization of atrigel
BLF and GF
The BLF and GF were measured using the universal testing machine (Model: UTM10, Make: Victoria Testing). The sample was mounted on the UTM machine using the tooling required to hold the syringe. The machine was set to the parameters, and the run was started using the maximum force of 80 N. The BKL and GF were recorded [29-31].
Drug loading analysis
An accurately weighed 5 ml of formulation, equivalent to 100 mg of GA, was transferred to a 100 ml volumetric flask. Dissolve the sample and dilute to volume with purified water. The solution was filtered through a 0.45 μm membrane filter. The solution was analysed using a UV-spectrophotometer (model UV-2600i plus, Shimadzu) at λmax at 275 nm [32].
Apparent viscosity
The apparent viscosity of the formed depot formulation was measured using a Brookfield viscometer (Model: DV2T). An accurately measured 20 ml formulation was charged into the sample holder. The liquid state solution was determined using an adapter: spindle T-bar T-A, spindle speed rpm: 20, 10-minute time, temperature 25 °C, and multi-average reading. The depot viscosity was determined using an adapter: spindle T-bar T-F (S-96), spindle speed rpm: 10, 10-minute time, temperature 37 °C, and endpoint reading. The readings were noted after finishing the run [33].
Scanning electron microscopy (SEM)
The morphology of the prepared formulations was analysed using SEM (model SU-3500, Hitachi). Briefly, dry, powdered particles were mounted on carbon-taped aluminium tubs and sputter-coated with gold using an argon evaporator under high vacuum. The samples were then observed using SEM [34, 35].
UV spectroscopy of GA
The UV spectrum of GA in a 7.4 pH phosphate buffer saline (PBS) was scanned from 400 nm to 200 nm using a 1 cm cell. The maximum absorbance was measured using a UV-Visible spectrophotometer (model UV-2600i plus, Shimadzu) to confirm the drug's maxima. The UV calibration curve was plotted against absorbance vs concentration [32, 36].
Fourier transform infrared spectroscopy (FTIR)
The chemical structure of the drug was analysed by using an attenuated total reflection (ATR) FTIR spectrometer (Model Make: Bruker II Alpha). The sample was kept at an ambient temperature of 25.0±0.5 °C. A few mg of the sample was placed on the zinc solenoid crystal plate; the anvil was rotated to fix the sample, and the spectra were recorded by scanning the samples in the region of 4000-400 cm-1 to determine various functional groups [37].
Differential scanning calorimetry (DSC)
A DSC scan (Model: DSC Model 250, TA Instrument) was performed to measure the thermograms of bulk GA, mannitol, Resomer 202H, the physical mixture, and the GA implant. Each sample was accurately weighed (2.0 mg) and charged into an aluminium pan with a puncture lid. A nitrogen gas flow rate of 50 ml/min was employed with a heating rate of 40-250 °C at 10 °C/min [38].
X-ray diffraction pattern (XRD)
X-ray diffraction patterns were recorded for GA, the polymer, and the final formulation to evaluate the physical nature of the formulations. XRD patterns were recorded on a Rigaku Smart Lab powder X-ray diffractometer at a scanning rate of 1° min−1 between 10° and 50° 2θ range [38].
pH measurement
The pH of the Atrigel formulation was measured using the pH meter (model: Orion Lab Star PH111, Thermo Fisher Scientific). The pH meter was calibrated at multiple pH values, and the Atrigel formulation was dispensed into the cuvette. The pH meter rod was immersed in the formulation, allowed to stabilise, and then recorded [39, 40].
Drug solubility study
The solubility of the GA was determined using the shake flask method. An excess amount of GA was added to 10 ml of PBS in a volumetric flask, and the flask was mounted on the shaker (model: Hei-shake orbital core 2000, Heidolph). After 24 h, the solution was filtered and analysed for the drug content. Additionally, the filtered solution was charged for 2-8˚C for 5 d to observe any recrystallisation [41].
In vitro dissolution study
The dissolution of bulk GA powder, Physical mixture (PM), GA-Atrigel was studied employing the USP Apparatus 2 (Model: Distek Inc., USA, North Brunswick) and Japanese sinkers. The paddle speed was monitored continuously at 50 rpm, and the temperature was maintained at 37 °C ±0.5 °C throughout the study. The solution was allowed to form a depot in a vial and then immersed in 30 ml of PBS. Aliquots of the receptor medium (1 ml) were withdrawn at predefined time intervals and replaced with fresh 1 ml buffer to maintain the sink condition. The concentration of the released drug in the samples was quantified using a validated analytical method, such as HPLC-UV, to generate the cumulative release profile over time [26, 34, 42].
Ex vivo release study of GA-atrigel
An ex vivo release study was conducted to evaluate intramuscular administration of the Atrigel formulation in the hen drumstick muscle, using previously published methodology. The hen drumstick model has been used previously and has demonstrated successful results. The porcine and rodent SC skin needs to be approved for use in the study. However, the hen drumstick is readily available [27]. The optimised formulation was injected subcutaneously into a hen drumstick muscle using a 20-gauge needle. The formulation-injected drumstick was immersed in a beaker containing 250 ml of pH 7.4 PBS supplemented with 0.1% w/v sodium azide as an antibacterial agent, and maintained at 37 °C throughout the study. The solution was stirred using a magnetic stirrer (model: Cimarec+™, Thermo Scientific). As the drug release study will be carried out over a more extended period, to prevent microbial growth in the solvent system, sodium azide has been used. The United States Food and Drug Administration (USFDA) recommends 0.1% w/v sodium azide, which is commonly used in IVPT studies to protect the media from microorganisms. Sodium azide is widely used in ex vivo skin permeation to protect the receptor media from microbial growth; it has been suggested and approved by the USFDA. It is widely used without interfering with the results. Multiple studies have used or recommended sodium azide in ex vivo studies to protect the skin or the target subject [43-45]. 1 ml of the release medium was taken out at predetermined intervals (2, 6, 8, 10, 12, 18, 24, 48, and 72 h) and replaced with an equal volume (1 ml) of fresh receptor medium to maintain sink conditions. To verify atrigel formation or implant formation, the injection site was incised 24 h after injection. The concentration of the released drug in the medium was analysed using high-performance liquid chromatography (HPLC). The cumulative drug release profile was subsequently evaluated based on the measured concentrations.
In vitro cytotoxicity assessment of formulations on L929 fibroblasts
The mouse L929 fibroblast cell line was cultured in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Cells were monitored at 37 °C in a humidified environment containing 5% CO2. The cells were seeded in 12-well plates at a density of 5 × 10⁴ cells per well and allowed to adhere for 24 h. Sterilised atrigel, implant formulations, NMP, and polymer (10 μl each sample) were applied directly to the cell monolayer. Usually, studies have been performed by extracting NMP from the formulation; however, this may affect the formulation's integrity and microstructure. Hence, this study explores the formulation without extraction. Additionally, the concentration of NMP in the sample and the standalone is different. Therefore, the direct formulation has been used rather than the extraction process. Additionally, the formulation without extraction reflects real-life conditions. The plates were incubated for a further 24 h. Untreated cells cultured in growth medium act as the negative control. Cell viability was determined employing the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The culture medium was aspirated and replaced with 500 μl of MTT solution (0.5 mg/ml in PBS). After a 4-hour incubation period, the MTT solution was withdrawn, and the formazan crystals were solubilised with 100 μl DMSO by shaking the plates for 1 hour. Absorbance was measured at 570 nm with a reference wavelength of 630 nm using a microplate reader (Tecan Group Ltd., Switzerland). All experiments were performed in triplicate, and results are presented as mean values±standard deviation [46].
RESULTS
Formulation optimization
DMSO, EA, and DCM were considered for formulation development but rejected in favour of NMP due to DMSO’s potential toxicity and drug stability concerns, ethyl acetate’s high volatility, which could lead to uncontrolled release, and DCM’s carcinogenic risk and regulatory restrictions. NMP was chosen for its moderate volatility, improved biocompatibility, and established use in FDA-approved depot formulations, ensuring safer, more controlled drug delivery in the Atrigel system. The trials GA-8 and GA-10 have the same composition and process, demonstrating reproducible results. This indicates the method and composition are reproducible.
ANOVA was used to evaluate the effects of variables and to select the model for response analysis, as shown in table 3. The independent factors X1 (Drug-polymer ratio) and X2 (solvent concentration) were treated as A and B, respectively, in the model. The independent factors A and B significantly affected GF, viscosity, and % release. The quadratic model best described the link between them.
Equation 1: Y1 = 6.47-2.03A+0.8637A+0.7597B-0.3967B
Equation 2: Y2 = 210900000– 5712.61A– 4630.06A+20081.06B-0.3349.78B+10406.94AB– 9452.06AB+1817.78AB+752.78AB
Equation 3: Y3 = 83.55+14.85A– 0.2789A– 9.62B– 2.96B+9.49AB– 9.77AB+3.44AB+7.53AB
A positive sign indicates that a factor improves the response, whereas a negative sign indicates that the relationship between the factor and the response is inverted. Equation 2 for viscosity includes a negative coefficient with respect to solvent concentration, suggesting that increasing solvent concentration typically lowers viscosity. The 3D response surface plot in the fig. illustrates how a factor affects the response. Learn more about the interrelationship between components A and B by using this plot. To understand how the various parameters affected the CQAs, the 3D surface plot was employed. Fig. 2 illustrates the effects of interactions between multiple formulations and composition parameters on GF, Viscosity, and % drug release.
Table 2: 32 Full factorial design and response values of GA formulation
| Batch no: | Factor 1 A: Drug: Polymer (mg) |
Factor 2 B: Solvent conc (mg) |
Response 1 GF [N] |
Response 2 depot viscosity [cP] | Response 3 % release [%] |
| GA-1 | 20:20 | 115 | 5.687±0.98 | 235674±5203 | 98.27±1.12 |
| GA-2 | 20:200 | 345 | 7.438±1.22 | 208036±6535 | 92.24±0.98 |
| GA-3 | 20:20 | 230 | 3.879±0.55 | 203654±2935 | 98.88±0.57 |
| GA-4 | 20:200 | 230 | 7.349±1.63 | 215324±3492 | 55.22±9.02 |
| GA-5 | 20:200 | 115 | 8.034±1.45 | 240368±5601 | 59.63±7.24 |
| GA-6 | 20:100 | 345 | 7.143±1.19 | 198236±3989 | 98.08±0.67 |
| GA-7 | 20:100 | 115 | 7.982±0.84 | 216897±6345 | 63.88±5.92 |
| GA-8 | 20:100 | 230 | 7.015±1.11 | 215319±4935 | 86.88±6.23 |
| GA-9 | 20:20 | 345 | 3.754±0.60 | 176230±2856 | 98.04±0.49 |
| GA-10 | 20:100 | 230 | 6.890±1.28 | 203671±4953 | 87.84±2.45 |
Results are given in mean±SD, n=3
Table 3: Analysis of variance (ANOVA) statistical model
| Source | GF (Y1) | Viscosity (Y2) | % Release (Y3) | ||||||||||||
| Sum of squares | df | Mean square | F-value | P-value | Sum of squares | df | Mean square | F-value | P-value | Sum of squares | df | Mean square | F-value | p-value | |
| Model | 20.51 | 8 | 2.56 | 20030.42 | 0.0055 | 3031000000 | 8 | 378800000 | 30310000 | 0.0001 | 3468.81 | 8 | 433.60 | 7501.75 | 0.0089 |
| A-Drug: Polymer | 17.45 | 2 | 8.72 | 68151.06 | 0.0027 | 524200000 | 2 | 262100000 | 20970000 | 0.0002 | 1710.84 | 2 | 855.42 | 14799.63 | 0.0058 |
| B-Solvent Conc | 2.69 | 2 | 1.35 | 10516.18 | 0.0069 | 2105000000 | 2 | 1053000000 | 84210000 | <0.0001 | 846.19 | 2 | 423.09 | 7319.97 | 0.0083 |
| AB | 1.07 | 4 | 0.2683 | 2095.74 | 0.0164 | 450800000 | 4 | 112700000 | 9017000 | 0.0002 | 738.27 | 4 | 184.57 | 3193.21 | 0.0133 |
| Residual | 0.0001 | 1 | 0.0001 | - | - | 12.5 | 1 | 12.5 | - | - | 0.0578 | 1 | 0.0578 | - | - |
| Lack of Fit | 20.51 | 9 | - | - | - | 3031000000 | 9 | - | - | - | 3468.87 | 9 | - | - | - |
| Pure Error | 20.51 | 8 | 2.56 | 20030.42 | 0.0055 | 3031000000 | 8 | 378800000 | 30310000 | - | 3468.81 | 8 | 433.60 | 7501.75 | 0.0089 |
| Cor Total | 17.45 | 2 | 8.72 | 68151.06 | 0.0027 | 524200000 | 2 | 262100000 | 20970000 | - | 1710.84 | 2 | 855.42 | 14799.63 | 0.0058 |
Note: df-degree of freedom
By comparing observed values with the predicted responses, we verified the correctness of the optimised formula. The experimental results were within the expected range, as shown in table 4. The relative error between the anticipated and observed results was less than 5%. To optimise the Atrigel depot formulations, a 32-way randomised complete factorial design was validated.
Table 4: Optimised formulation compositions and predicted vs. observed response values
| Optimized formulation | Drug: polymer ratio | Content of solvent per unit dose | |
| Optimized concentration | 0.97222 | 230 | |
| Responses | GF (N) | Viscosity [depot] (cP) | % Release |
| Predicted value | 7.249 | 215321 | 55.05 |
| Observed value | 7.255 | 215270 | 53.98 |
| % Relative error | 0.083 | 0.003 | -1.944 |
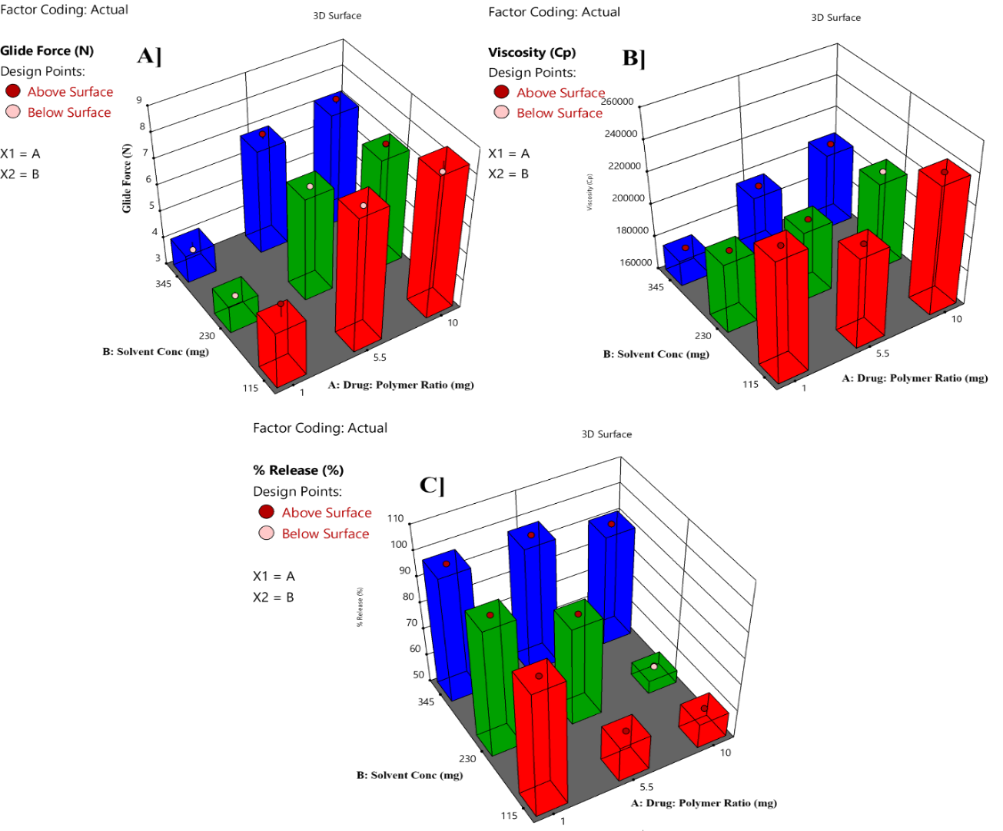
Fig. 2: 3D response surface plot representing the interpretation of independent variables on A] GF, B] Viscosity, C] % Release Results for drug loading, BLF, pH, and viscosity pre-injection solution state are recorded in table 5
Table 5: Results for drug loading, BLF, pH, and viscosity [Pre-injection]
| Batch No: | % Drug loading | BLF (N) | pH | Viscosity (cP) |
| GA-1 | 99.2±0.45 | 11.825±0.25 | 6.50±0.05 | 1250±12.4 |
| GA-2 | 99.9±0.22 | 10.265±0.55 | 7.56±0.02 | 1103±14.5 |
| GA-3 | 99.3±0.49 | 5.205±0.61 | 7.03±0.07 | 1080±13.7 |
| GA-4 | 99.7±0.52 | 12.219±0.85 | 7.25±0.06 | 1142±11.4 |
| GA-5 | 99.8±0.89 | 11.650±0.54 | 6.62±0.05 | 1275±14.8 |
| GA-6 | 99.5±0.67 | 11.595±0.87 | 7.26±0.03 | 1051±17.2 |
| GA-7 | 99.4±0.36 | 15.794±0.34 | 6.83±0.04 | 1150±15.8 |
| GA-8 | 99.9±0.53 | 12.501±0.50 | 7.31±0.07 | 1138±16.2 |
| GA-9 | 99.1±0.73 | 6.507±0.58 | 7.35±0.06 | 935±16.3 |
| GA-10 | 99.6±0.56 | 8.559±0.74 | 7.23±0.08 | 1080±13.0 |
Results are given in mean±SD, n=3
Characterization of atrigel
Drug loading analysis
Results showed that drug loading across all ten batches (GA-1 to GA-10) consistently ranged from 99.1% to 99.9%, with minimal standard deviation (all below 0.9%). This demonstrates that, within the tested process parameters, the final product achieved near-complete loading efficiency with excellent batch-to-batch consistency. Firstly, changes in the drug-to-polymer ratio (for example, increasing the polymer quantity from equal to ten times the drug amount) did not significantly impact loading efficiency. Both high loading ratios (20:20) and low ratios (20:200) produced efficiencies approaching 100%. This reflects the polymer's strong drug-encapsulation capacity, remaining well below its saturation limit even at the lowest ratio tested (20:200). Secondly, variations in solvent concentration across three levels did not cause systematic changes in drug loading. Consistently high drug-loading results were obtained across low, medium, and high solvent concentrations, indicating that variations in solvent usage do not affect the key step of effective drug encapsulation by the polymer in the current process system.
BLF and GF
The BLF, an indicator of the force required to expel the gel from the syringe, showed the widest variation, with values ranging from 6.5071~15.794 N. Formulations with a higher drug-to-polymer ratio of 20:200 consistently resulted in a much higher BLF around 10.2~12.5N compared to the lower ratio of 20:20, which had an average BLF of only 1.62 N.
Apparent viscosity
The solution-state viscosities are lower than the depot, ranging from approximately 935 to 1275 centipoise (cP), indicating the solution is injectable. Contrarily, the solution after depot formation is a viscosity enhancement. The viscosity after deposition formation lies between 176,230~240,368 cP, which is roughly 200 times greater than their solution counterparts. The variability in solution and depot viscosity is proportionally similar to that of the solutions.
Overall, the drug-to-polymer ratio had a greater impact on the gel's physical properties than the solvent concentration. Drug-polymer ratio and solvent concentration both play an essential role in Atrigel formulations because they control system stability and the drug release profile. The two criteria above showed a considerable impact on the GA-Atrigel formulation. Hence, a 32 randomised complete factorial design with one replicate point was employed to evaluate the essential elements for optimisation: the solvent concentration and the drug-to-polymer ratio in the formulation. In this investigation, there were nine experimental runs and one replicate point. Table 3 summarises the outcomes of the selected response for all experiments, including GF, viscosity, and in vitro release. Various formulation batches of GA Atrigel were prepared based on the factorial design. The independent variables were drug-to-polymer ratio (X1) and Solvent concentration (X2).
SEM
The microstructural analysis demonstrated three distinct morphological profiles at scale bars of 50.0, 10.0, and 10.0 μm for GA, the physical mixture, and the polymer PLGA, respectively, as shown in fig. 3. The GA has a crystalline structure, exhibiting angular facets and sharp edges. The polymer particles are irregular aggregates with a porous surface. Physically homogeneous mixtures exhibit intermediate characteristics. Notably, the GA (GA)-incorporated Atrigel systems consistently yielded smooth matrices with submicron surface roughness (Ra<100 nm), as quantified using image analysis software. GA's appearance is very soft, whereas the physical mixtures appear uniform, with no visible polymer or drug.

Fig. 3: SEM of A] GA; B] Physical mixture; C] Polymer PLGA SEM image
UV calibration curve of GA
The UV analysis of GA in PBS, pH 7.4, revealed a λmax at 275 nm, consistent with the chromophoric properties of GA's polypeptide structure, as shown in fig. 4B]. The calibration curve has demonstrated excellent linearity (R² = 0.998) when constructed by plotting absorbance versus GA concentration (range: 0-80 μg/ml). The curve exhibited confirming adherence to the Beer-Lambert law, as shown in fig. 4A]. This validated linear relationship demonstrates the method's suitability for accurate GA quantification in dissolution testing and in vitro release studies.
FTIR
FTIR spectral analysis of the physical mixture demonstrated characteristic absorption bands corresponding to both components: the amide I (∼1650 cm⁻¹), amide II (∼1540 cm⁻¹), and carboxylic group (~3298 cm-1) vibrations of GA's peptide backbone, along with the ester carbonyl stretch (∼1750 cm⁻¹) of PLGA. The results demonstrate that the preservation of the molecular integrity of GA, without significant chemical interactions during formulation processing, as shown in fig. 5. The FTIR spectra revealed characteristic peaks of GA (amide I at ~1650 cm⁻¹) and PLGA (ester C=O at ~1750 cm⁻¹). Although these peaks overlap, no new absorption bands were observed, confirming the absence of covalent interactions between GA and PLGA and indicating chemical stability in the formulation. This suggests that the drug-polymer interaction is primarily physical, supporting the integrity of the Atrigel system.
DSC
DSC was employed to evaluate the thermal behaviour and compatibility of GA with the PLGA polymer matrix. The thermograms demonstrated distinct endothermic transitions corresponding to the melting behaviour of crystalline GA and the glass transition of amorphous PLGA, as shown in fig. 6. Analysis of the physical mixture revealed modified thermal events, including shifts in transition temperatures and changes in enthalpy values, and maintained the characteristic thermal signatures of both components. The preserved thermal properties of both elements in the mixture further support the maintenance of their structural integrity and the formation of a physically stable composite matrix.
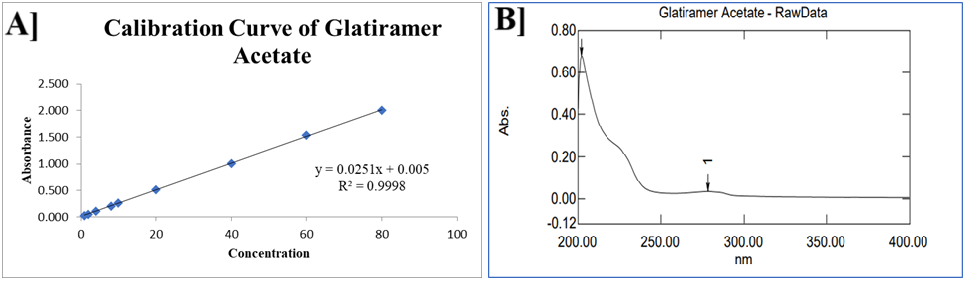
Fig. 4: A] UV Calibration curve; B] λmax of GA
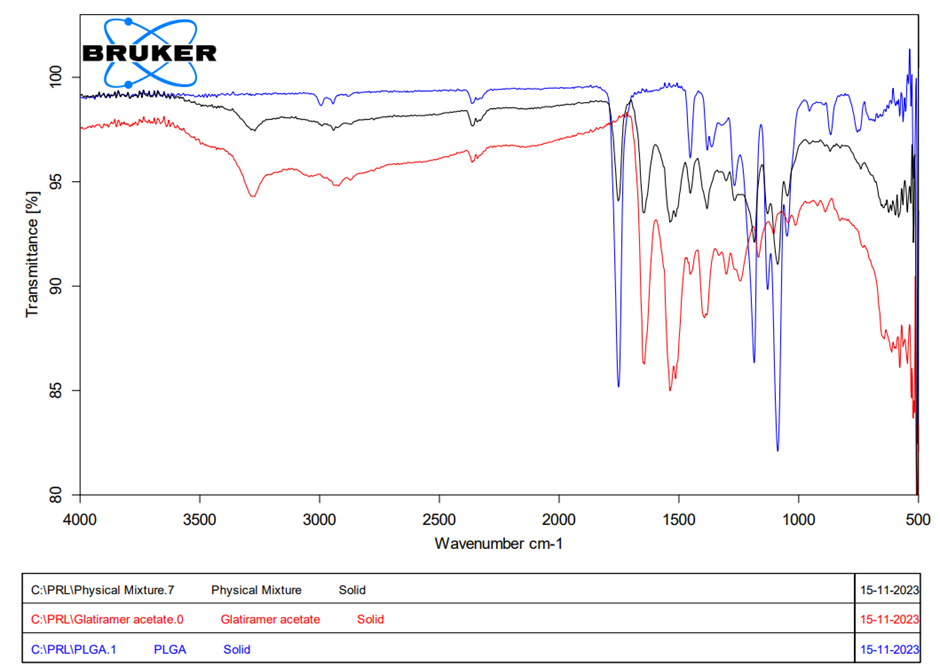
Fig. 5: FTIR spectra of GA, physical mixture and polymer
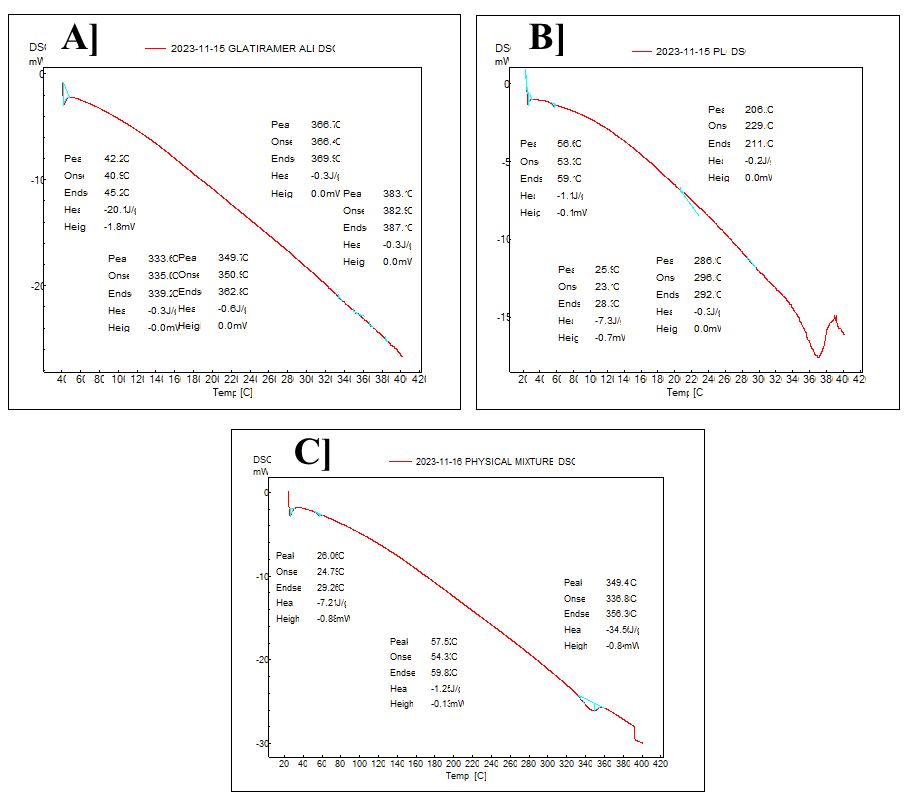
Fig. 6: DSC thermogram of A] GA; B] PLGA polymer; C] Physical mixture
XRD
The X-ray diffraction pattern of pure GA displayed distinct Bragg reflections at characteristic 2θ angles, confirming its crystalline nature, as shown in fig. 7. Conversely, the Atrigel formulations exhibited a significant attenuation of these crystalline peaks, along with the emergence of a broad halo pattern. This remarkable reduction in crystallinity indicates the successful molecular dispersion of GA into the polymer matrix, with the drug transitioning to an amorphous state, which is expected to enhance dissolution and controlled-release kinetics.
pH measurement
The pH of the formulations was generally neutral to slightly basic, with values spanning from 6.50 to 7.56.
Drug solubility study
GA is a water-soluble copolypeptide with a solubility of approximately 1 mg/ml in PBS at pH 7.4. Based on this, it can be concluded that the receptor media won’t be saturated until the drug is completely released, as 30 ml of receptor media has been used.
In vitro dissolution study
GA, drug-polymer mixture of GA (Physical Mixture-PM), and the optimised formulation dissolution profile were studied in 7.4 PBS, and the results are graphically represented in fig. 8A. PM shows a slight decrease in the dissolution profile over time compared to GA. The cumulative dissolution of PM was 100% in 75 h, whereas for GA it was 20 h. In situ-forming GA implant showed a 55% cumulative dissolution in 120 h. In a pH 7.4 PBS, the similarity factor f2 for the dissolution profiles of GA was 26, and f1 is likewise less than for GA implant and PM. In situ implant demonstrated significantly superior controlled release than GA and PM. The extended release of the GA implant's dissolution rate was therefore inferred to indicate good drug delivery, which will definitely increase its half-life and extend its duration of action, subsequently decreasing the dose frequency. The drug release kinetic study data are represented in table 6. Based on the coefficient of determination (R²), the models' goodness of fit, ranked from highest to lowest, is as follows: Higuchi model (0.9842)>first-order kinetic model (0.9747)>Hixson-Crowell model (0.9613)>zero-order kinetic model (0.9261)>Korsmeyer-Peppas model (0.8748). The Higuchi model has the highest R2 value; hence, the dosage form follows the Higuchi drug release pattern.
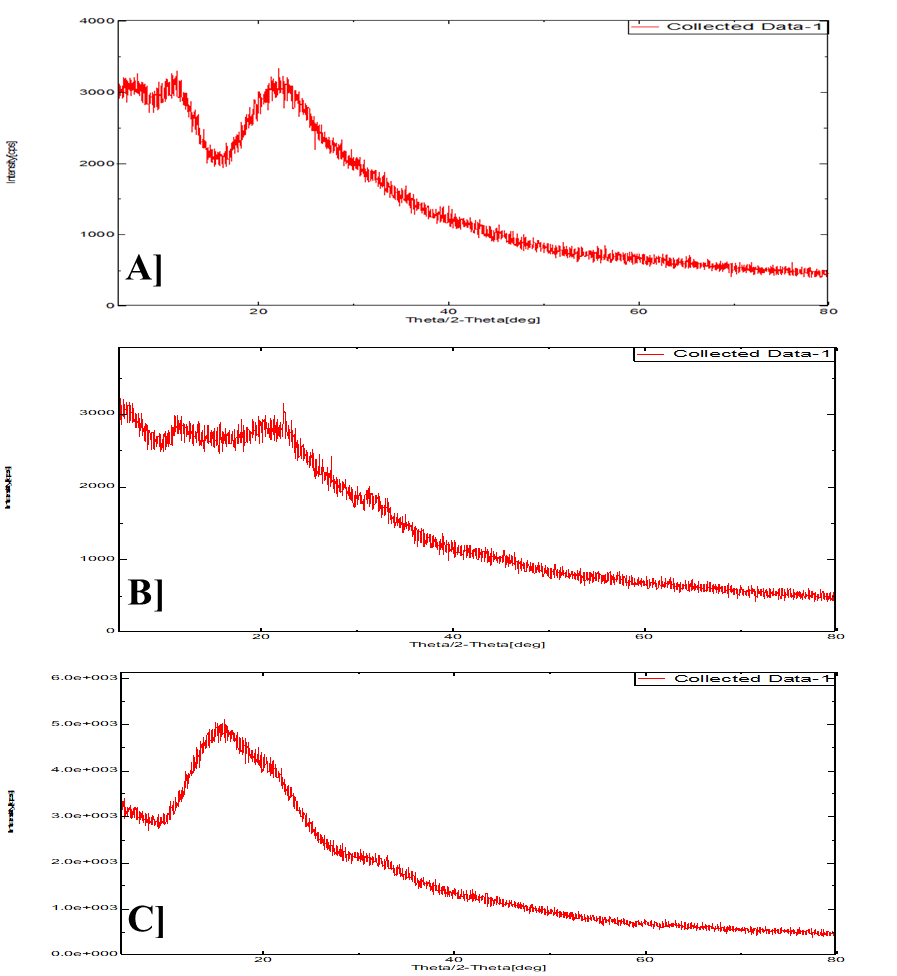
Fig. 7: XRD spectra of A] GA; B] Atrigel formulation; C] PLGA polymer
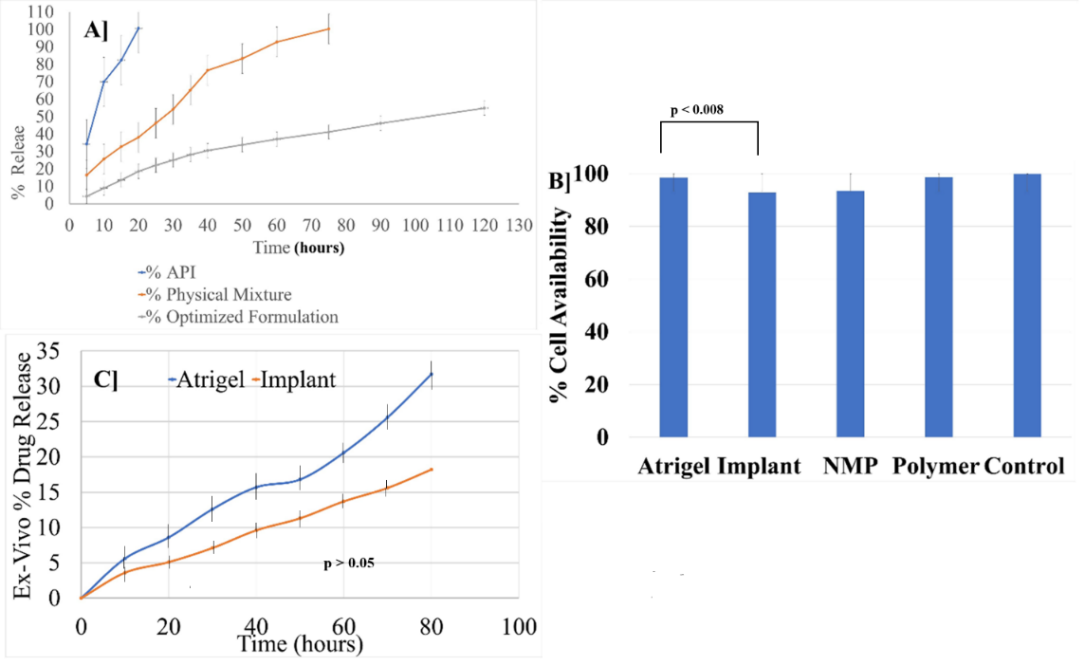
Fig. 8: Results A] In vitro % drug release; B] % cell availability; C] Ex vivo % drug release. Error bars indicated SD values
Table 6: In vitro drug release kinetics study
| Model | R2 | Slope | Intercept |
| Zero order | 0.9261 | 0.4441 | 7.8294 |
| Fist-order | 0.9747 | -0.0028 | 1.9739 |
| Huguchi model | 0.9842 | 5.3858 | -4.8536 |
| Hixson | 0.9613 | 0.0088 | 0.1056 |
| Kor's peppas | 0.8748 | 27.165 | -11.93 |
Ex vivo evaluation of GA-atrigel
The ex vivo study was conducted to assess the formation and morphology of atrigels and implants (PLGA 504H) and to evaluate the release kinetics of the active GA from hen drumstick tissue. To confirm depot formation, the injected formulations were examined 24 h post-administration by dissecting the drumstick tissue, as illustrated in fig. 8C; the red frames highlight the subcutaneous deposition of the implant and gel, respectively. Ex vivo drug release was monitored over 72 h, after which the release medium exhibited signs of degradation. Comparative analysis of the release profiles from in situ-forming gels and in situ-forming implants revealed no statistically significant differences (p>0.05) between ex vivo and in vitro release kinetics, confirming consistent behaviour upon subcutaneous administration.
In vitro in vivo correlation (IVIVC)
The in vitro-in vivo correlation (IVIVC) for the Atrigel formulation was established based on a comparative analysis of in vitro dissolution and ex vivo release studies. The in vitro dissolution study in pH 7.4 PBS revealed that the GA in situ forming implant exhibited a controlled release profile, with only 55% cumulative drug release over 120 h, significantly slower than the PM and pure GA, which released 100% within 75 h and 20 h, respectively. It can be attributed that the complete drug release will happen in around ~218 h, approximately 9 d. Therefore, a single dose of the Atrigel formulation delivers the drug for about 9 d. The similarity factors (f2 = 26, f1<15) further confirmed the distinct release kinetics of the implant, suggesting its potential for extended drug delivery. The ex vivo study in hen drumstick tissue demonstrated consistent subcutaneous depot formation, with no significant difference (p>0.05) between in vitro and ex vivo release profiles over 72 h, indicating reliable in vivo predictability. These findings suggest a strong correlation between in vitro dissolution and ex vivo release, supporting the Atrigel formulation's ability to sustain drug release, prolong half-life, and reduce dosing frequency in vivo.
In vitro cytotoxicity assessment on L929 fibroblasts
The adipose tissue-derived mouse L929 fibroblast cell line was selected for cytotoxicity testing due to its relevance to subcutaneous connective tissue. Fibroblasts are a prominent cell type in the dermal layer and serve as a suitable model for assessing biocompatibility. The mitochondrial dehydrogenase activity in viable cells was determined via the reduction of MTT to formazan for cytotoxicity using the MTT assay. The fibroblast viability after 24 h of exposure to the formulations ranged 92.8~99.9%, confirming acceptable biocompatibility, as shown fig. 8B. The Atrigel sample demonstrated significantly higher cell viability (98.5±1.15%) than the implant (92.8±2.93%) (p<0.008), attributed to reduced NMP burst release over the initial 24 h. The current research findings align with in vitro release data, supporting the conclusion that the Atrigel formulation system is biocompatible and suitable for subcutaneous use.
DISCUSSION
The development of a long-acting Atrigel® formulation of GA for MS therapy represents a significant advancement in addressing treatment adherence challenges [48-50]. Our optimised formulation exhibits controlled release kinetics, with only 55% cumulative drug release, while bulk GA occurs within 20 min. This sustained-release profile could be attributed to optimising the drug-to-polymer ratio and solvent concentration, suggesting the potential to reduce dose frequency while maintaining therapeutic efficacy. The factorial design approach demonstrated a correlation between formulation variables and key performance attributes, with higher polymer ratios effectively modulating release rates. These findings align with previously established principles of PLGA-based depot systems, where polymer composition and solvent selection significantly influence drug release patterns [51, 52]. Equation 2 for viscosity includes a negative coefficient for solvent concentration, which means that increasing solvent concentration typically lowers viscosity. This could be mainly attributed to the solvent NMP. NMP is a strong solvent that reduces the apparent viscosity of the formulation by weakening bonds between polymer chains. The viscosity results show a pronounced and consistent increase after converting the liquid solution into a depot form. The viscosity of the solution and deport is 200 times greater. This significant rise in the viscosity of all formulation batches confirms that the gelation process is a fundamental property of the material that reliably produces a highly viscous solid implant. The table effectively quantifies the critical rheological transformation of the Atrigel system from a low-viscosity solution to a high-viscosity depot, a key characteristic for its application as an injectable, in-situ-forming implant [53, 54].
The BLF indicates the force required to start the plunger moving from its original position. In contrast, the gliding force indicates the additional force needed to push the plunger further and expel the solution from the syringe. These are essential aspects of the formulation's syringability, and the current formulation meets the basic criteria.
Comprehensive physicochemical characterisation confirmed the formulation's structural integrity and performance. SEM and XRD results demonstrated the transition of crystalline GA to an amorphous state within the PLGA matrix. DSC and FTIR results demonstrated chemical compatibility among the ingredients, with no chemical interactions. The developed UV spectroscopic method exhibited excellent linearity, enabling reliable quantification of the drug. In this study, the performance of the drug-to-polymer ratio and solvent concentration was investigated and evaluated. It was found that the Atrigel system has a significant impact on drug release compared to the simple injection of GA. A polymer exhibiting good stability in NMP is found to have better physical characteristics in formulation compared to other solvents.
Sodium azide has been widely used in in vitro release and permeation studies, and it has been assumed not to interfere with the analysis. It has also been used in an Atrigel formulation for 30-day research to protect the media. Other bacterial agents are not recommended for the in vitro studies [44, 45].
Biological evaluations additionally evidenced Atrigel’s potential. The drumstick model, with an ex vivo study, showed a release profile comparable to the in vitro results, validating the formation of a depot. Cytotoxicity assessments using L929 fibroblasts revealed excellent biocompatibility (97.8-100% viability), particularly for the Atrigel formulation (98.5±1.15%), consistent with previous reports on similar delivery systems [27, 51, 52]. Prior studies demonstrated that in situ-formulation implant systems exhibit fibroblast viability in the range of 96~100%, indicating minimal cytotoxicity [55, 56]. The extraction of NMP from the formulation affects formulation integrity through microstructural changes. Hence, this study explores the formulation without extraction. Additionally, the concentration of NMP in the sample and the standalone is different. Therefore, the direct formulation has been used rather than the extraction process.
The Higuchi model's R² value is closest to 1, indicating it most accurately describes the drug release process. This suggests the release mechanism is likely diffusion-controlled, where drug diffusion through the matrix or scaffold constitutes the rate-limiting step—a characteristic consistent with many hydrophilic or hydrophobic polymer systems. The zero-order kinetic model indicates a constant drug release rate over a specific time period. However, its fit is slightly inferior to that of the Higuchi model, suggesting that pure zero-order release characteristics do not entirely govern the process. The first-order kinetic model exhibits a negative slope, and a high R² value indicates a strong exponential relationship between drug release and residual drug quantity, with the release rate diminishing over time. This aligns with phenomena commonly associated with diffusion or erosion mechanisms. The Higuchi model's high slope and negative intercept further corroborate the strong linear relationship between drug release and the square root of time, providing compelling evidence for classically diffusion-controlled release. The Hixson-Crowell model exhibits a smaller slope, suggesting slow changes in formulation size or surface area over time. The Korsmeyer-Peppas model suggests limitations in applying this model to this specific system.
The IVIVC indicates that the 100% drug could be released in 9 d. Hence, we can expect a minimum of 7 d of drug release. The clinical application of the current research is substantial, which offers a potential solution to the patient compliance challenges associated with daily GA injections. The sustained release characteristics may enable less frequent dosing while maintaining therapeutic drug levels, similar to advanced long-acting injectables. The amorphous nature of GA in the Atrigel formulation will promote solubility and bioavailability, potentially allowing for dose reduction. However, more evidence is needed to support the claim of higher bioavailability and dose reduction. Atrigel platform technology is not only applicable to GA delivery but also potentially to other MS therapeutics requiring sustained-release profiles.
CONCLUSION
This study successfully demonstrated the development and optimisation of a GA Atrigel® drug delivery system for the management of multiple sclerosis. Using a systematic approach with a 32-randomised complete factorial design, the impact of key formulation parameters, such as drug-to-polymer ratio and solvent concentration, was evaluated. The Atrigel system exhibited superior performance in terms of drug encapsulation, stability, and controlled release compared to traditional formulations. Comprehensive characterisation using SEM, UV-spectroscopy, DSC, XRD, and FTIR confirmed the physical and chemical integrity of the formulation. In vitro dissolution studies demonstrated extended drug release profiles, which are expected to enhance therapeutic efficacy while reducing dosing frequency, as shown in ex vivo and in vivo studies. Results indicate that the Higuchi model is the best fit for this drug release system, confirming diffusion-controlled release. The first-order kinetic model also demonstrates a good fit, further supporting the diffusion-like characteristic of decreasing release rates over time. Conversely, the zero-order, Hixson-Crowell, and Korsmeyer-Peppas models exhibited comparatively lower fitting accuracy, indicating that purely constant-rate release, pure erosion mechanisms, or simple power-law relationships cannot fully describe the complex behaviour of this system. These results highlight the Atrigel system's potential as a promising alternative to conventional daily injections, offering improved patient compliance and extended action.
ACKNOWLEDGEMENT
The author expresses their sincere gratitude to Pacific Academy of Higher Education and Research University, Udaipur, Rajasthan, India, for their support and for providing the facilities for this research work. Lastly, we are grateful to all colleagues and collaborators who have provided valuable suggestions and assistance throughout the study.
AUTHORS CONTRIBUTIONS
Sushant Pote: Conceptualisation, data curation, formal analysis, investigation, methodology, resources supervision, validation, visualisation, writing/original draft preparation, review, and editing. Om Shelke: Conceptualisation, data curation, formal analysis, investigation, methodology, resources supervision, validation, visualisation, writing/original draft preparation, review, and editing. Rajesh Khathuriya: Conceptualisation, data curation, formal analysis, methodology, and draft review. Vrushali Kulkarni: Conceptualisation, data curation, formal analysis, methodology, and draft review. Sachin Fartade: conceptualisation, data curation, formal analysis, methodology, and draft review.
CONFLICT OF INTERESTS
The authors declare that they have no conflicts of interest related to this study or its publication.
REFERENCES
Compston A, Coles A. Multiple sclerosis. Lancet. 2002 Apr 1;359(9313):1221-31. doi: 10.1016/s0140-6736(02)08220-x, PMID 11955556.
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000 Sep 28;343(13):938-52. doi: 10.1056/nejm200009283431307, PMID 11006371.
Ruggieri M, Avolio C, Livrea P, Trojano M. Glatiramer acetate in multiple sclerosis: a review. CNS Drug Rev. 2007;13(2):178-91. doi: 10.1111/j.1527-3458.2007.00010.x, PMID 17627671.
Montague T, Drummond J, Ng K, Parratt J. Advancements in multiple sclerosis. Intern Med J. 2025;55(6):895-904. doi: 10.1111/imj.70023, PMID 40171883.
Ghasemi N, Razavi S, Nikzad E. Multiple sclerosis: pathogenesis symptoms diagnoses and cell-based therapy. Cell J. 2016;19(1):1-10. doi: 10.22074/cellj.2016.4867, PMID 28367411.
Ziemssen T. Modulating processes within the central nervous system is central to therapeutic control of multiple sclerosis. J Neurol. 2005 Oct 1;252(Suppl 5):v38-45. doi: 10.1007/s00415-005-5007-2, PMID 16254701.
Goldberg LD, Edwards NC, Fincher C, Doan QV, Al-Sabbagh A, Meletiche DM. Comparing the cost-effectiveness of disease-modifying drugs for the first-line treatment of relapsing-remitting multiple sclerosis. J Manag Care Pharm. 2009 Sep 1;15(7):543-55. doi: 10.18553/jmcp.2009.15.7.543, PMID 19739877.
Dobson R, Giovannoni G. Multiple sclerosis a review. Eur J Neurol. 2018 Oct 10;26(1):27-40. doi: 10.1111/ene.13819, PMID 30300457.
Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S. Multiple sclerosis. Nat Rev Dis Primers. 2018 Nov 8;4(1):43. doi: 10.1038/s41572-018-0041-4, PMID 30410033.
Rodriguez M. Diagnosis and management of multiple sclerosis. J Neuropathol Exp Neurol. 2003 May 1;62(5):590. doi: 10.1093/jnen/62.5.590.
Hahn JS, Pohl D, Rensel M, Rao S, International Pediatric MS Study Group. Differential diagnosis and evaluation in pediatric multiple sclerosis. Neurology. 2007;68(16 Suppl 2):S13-22. doi: 10.1212/01.wnl.0000259403.31527.ef, PMID 17438234.
Gelfand JM. Multiple sclerosis: diagnosis differential diagnosis and Clinical Presentation. Handb Clin Neurol. 2014 Jan 1;122:269-90. doi: 10.1016/b978-0-444-52001-2.00011-x, PMID 24507522.
Ciotti JR, Cross AH. Disease-modifying treatment in progressive multiple sclerosis. Curr Treat Options Neurol. 2018 Apr 7;20(5):12. doi: 10.1007/s11940-018-0496-3, PMID 29627873.
Oh J, Bar-Or A. Emerging therapies to target CNS pathophysiology in multiple sclerosis. Nat Rev Neurol. 2022 Jun 13;18(8):466-75. doi: 10.1038/s41582-022-00675-0, PMID 35697862.
Gholamzad M, Ebtekar M, Ardestani MS, Azimi M, Mahmodi Z, Mousavi MJ. A comprehensive review on the treatment approaches of multiple sclerosis: currently and in the future. Inflam Res. 2018 Sep 3;68(1):25-38. doi: 10.1007/s00011-018-1185-0, PMID 30178100.
Rocco P, Eberini I, Musazzi UM, Franze S, Minghetti P. Glatiramer acetate: a complex drug beyond biologics. Eur J Pharm Sci. 2019;133:8-14. doi: 10.1016/j.ejps.2019.03.011, PMID 30902653.
Lalive PH, Neuhaus O, Benkhoucha M, Burger D, Hohlfeld R, Zamvil SS. Glatiramer acetate in the treatment of multiple sclerosis: emerging concepts regarding its mechanism of action. CNS Drugs. 2011 Mar 30;25(5):401-14. doi: 10.2165/11588120-000000000-00000, PMID 21476611.
Teva Neuroscience, Inc. Copaxone (glatiramer acetate) injection for subcutaneous use. Parsippany (NJ): U.S. Food and Drug Administration; 2024. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/020622s118s119lbl.pdf. [Last accessed on 07 Jan 2026].
Varkony H, Weinstein V, Klinger E, Sterling J, Cooperman H, Komlosh T. The glatiramoid class of immunomodulator drugs. Expert Opin Pharmacother. 2009 Feb 26;10(4):657-68. doi: 10.1517/14656560902802877, PMID 19245345.
Schrempf W, Ziemssen T. Glatiramer acetate: mechanisms of action in multiple sclerosis. Autoimmun Rev. 2007;6(7):469-75. doi: 10.1016/j.autrev.2007.02.003, PMID 17643935.
Aharoni R, Milo R, Arnon R. Glatiramer acetate for the treatment of multiple sclerosis: from first-generation therapy to elucidation of immunomodulation and repair. Pharmacol Rev. 2024 Oct 15;76(6):1133-58. doi: 10.1124/pharmrev.124.000927, PMID 39406508.
Mirabella M, Annovazzi P, Brownlee W, Cohen JA, Kleinschnitz C, Wolf C. Treatment challenges in multiple sclerosis a continued role for glatiramer acetate? Front Neurol. 2022 Apr 15;13:844873. doi: 10.3389/fneur.2022.844873, PMID 35493825.
Denis L, Namey M, Costello K, Frenette J, Gagnon N, Harris C. Long-term treatment optimization in individuals with multiple sclerosis using disease-modifying therapies: a nursing approach. J Neurosci Nurs. 2004 Feb 1;36(1):10-22. doi: 10.1097/01376517-200402000-00003, PMID 14998102.
Flechter S, Popper L, Kimelman NB, Rubnov S, Danon U, Marom E. Glatiramer acetate depot (extended-release) phase iia study in patients with primary progressive multiple sclerosis: safety and efficacy snapshot (P7-4.001). Neurology. 2022;98(18_Supp):P7‑4.001. doi: 10.1212/WNL.98.18_supplement.1329.
Thakur RR, McMillan HL, Jones DS. Solvent-induced phase inversion-based in situ forming controlled release drug delivery implants. J Control Release. 2014 Jan 2;176:8-23. doi: 10.1016/j.jconrel.2013.12.020, PMID 24374003.
Park K, Otte A, Sharifi F, Garner J, Skidmore S, Park H. Formulation composition manufacturing process and characterization of poly(lactide-co-glycolide) microparticles. J Control Release. 2020 Oct 24;329:1150-61. doi: 10.1016/j.jconrel.2020.10.044, PMID 33148404.
Kamali H, Khodaverdi E, Hadizadeh F, Mohajeri SA. In vitro, ex-vivo, and in vivo evaluation of buprenorphine HCl release from an in situ forming gel of PLGA-PEG-PLGA using N–methyl–2-pyrrolidone as solvent. Mater Sci Eng C Mater Biol Appl. 2018 Nov 28;96:561-75. doi: 10.1016/j.msec.2018.11.058, PMID 30606566.
Avachat AM, Kapure SS. Asenapine maleate in situ forming biodegradable implant: an approach to enhance bioavailability. Int J Pharm. 2014 Oct 9;477(1-2):64-72. doi: 10.1016/j.ijpharm.2014.10.006, PMID 25305379.
Primavessy D, Piening M, Nightingale A, Jameson H, Latham M, Alexander J. Investigation of long-term pressure on primary packaging materials and a biologic drug product for injection with a novel autoinjector concept. Drug Deliv Transl Res. 2024;15(2):577-95. doi: 10.1007/s13346-024-01612-y, PMID 38727986.
Alkeefo R, Hotz C, Kolacyak D. Impact of dimensional variability of primary packaging materials on the break-loose and gliding forces of prefilled syringes. PDA J Pharm Sci Technol. 2024;78(5):572-85. doi: 10.5731/pdajpst.2023.012916, PMID 39164114.
Shi GH, Gopalrathnam G, Shinkle SL, Dong X, Hofer JD, Jensen EC. Impact of drug formulation variables on silicone oil structure and functionality of prefilled syringe system. PDA J Pharm Sci Technol. 2017;72(1):50-61. doi: 10.5731/pdajpst.2017.008169, PMID 29030531.
Hadidi N, Pazuki G. Preparation, characterization and in vivo efficacy study of glatiramer acetate (GA)-hydrogel-microparticles as novel drug delivery system for GA in RRMS. Sci Rep. 2022;12(1):22042. doi: 10.1038/s41598-022-26640-x, PMID 36543898.
Liu Q, Zhang H, Zhou G, Xie S, Zou H, Yu Y. In vitro and in vivo study of thymosin alpha 1 biodegradable in situ forming poly(lactide-co-glycolide) implants. Int J Pharm. 2010 Jul 21;397(1-2):122-9. doi: 10.1016/j.ijpharm.2010.07.015, PMID 20650309.
Molavi F, Barzegar Jalali M, Hamishehkar H. Changing the daily injection of glatiramer acetate to a monthly long-acting product through designing polyester-based polymeric microspheres. Bio Impacts. 2022 Aug 13;12(6):501-13. doi: 10.34172/bi.2022.23733, PMID 36644544.
Hadidi N, Pazuki G. Preparation, characterization and in vivo efficacy study of glatiramer acetate (GA)-hydrogel-microparticles as novel drug delivery system for GA in RRMS. Sci Rep. 2022 Dec 21;12(1):22042. doi: 10.1038/s41598-022-26640-x, PMID 36543898.
Rahiman N, Zamani P, Arabi L, Alavizadeh SH, Nikpoor A, Mashreghi M. Novel liposomal glatiramer acetate: preparation and immunomodulatory evaluation in murine model of multiple sclerosis. Int J Pharm. 2023 Nov 18;648:123620. doi: 10.1016/j.ijpharm.2023.123620, PMID 37981250.
Hajian M, Erfani Moghadam V, Arabi MS, Soltani A, Shahbazi M. A comparison between optimized PLGA and CS-Alg-PLGA microspheres for long-lasting release of glatiramer acetate. J Drug Deliv Sci Technol. 2023 Mar 10;82:104355. doi: 10.1016/j.jddst.2023.104355.
Salarieh Z, Esmaeili A, Pad MH. Synthesis of cubosomes containing cerium oxide nanoparticles from Lactobacillus acidophilus loaded with glatiramer acetate and carboxymethylcellulose coating. Int J Biol Macromol. 2023;231:123215. doi: 10.1016/j.ijbiomac.2023.123215, PMID 36642361.
Jie F, Shelke O, Yijie Z, Yulan C, Yongbo L. Q1 and Q2 selection, Q3, IVRT, IVPT, pharmacokinetic and pharmacodynamic evaluation of topical generic product. Drug Dev Ind Pharm. 2025 Apr 4;51(6):555-65. doi: 10.1080/03639045.2025.2486487, PMID 40176255.
Shelke O, Susarla KP, Bankar M. Understand the stabilization engineering of ascorbic acid mapping the scheme for stabilization and advancement. AAPS PharmSciTech. 2024 Jul 11;25(6):159. doi: 10.1208/s12249-024-02882-y, PMID 38987438.
Padervand M, Elahifard MR. Development of a spectrophotometric method for the measurement of kinetic solubility: economical approach to be used in pharmaceutical companies. Pharm Chem J. 2017 Sep 1;51(6):511-5. doi: 10.1007/s11094-017-1645-9.
Farahmand D, Mehrabi MR, Eidi A. Enhancing drug delivery of glatiramer acetate through in vitro development of controlled-release nanoliposomes: investigating drug release kinetics and cytotoxic effects. Jundishapur J Nat Pharm Prod. 2024 Sep 3;19(4):e145855. doi: 10.5812/jjnpp-145855.
U.S. Food and Drug Administration (FDA). Guidance for industry: in vitro permeation test studies for topical drug products submitted in ANDAs. Center for Drug Evaluation and Research, Office of Regulatory Policy; 2022. FDA-2022-D-1862. Available from: https://www.fda.gov/media/162475/download.
Patel HA. Practical considerations related to in vitro permeation test studies for topical products submitted in ANDAs. In: SBIA 2022: Best practices for topical generic product development and ANDA submission. Virtual public webinar. Silver Spring (MD): Office of Bioequivalence, Office of Generic Drugs, CDER, FDA; 2022 Aug. Available from: https://www.fda.gov/media/165074/download. [Last accessed on 07 Jan 2026].
Agonia AS, Palmeira De Oliveira A, Cardoso C, Augusto C, Pellevoisin C, Videau C. Reconstructed human epidermis: an alternative approach for in vitro bioequivalence testing of topical products. Pharmaceutics. 2022;14(8):1554. doi: 10.3390/pharmaceutics14081554, PMID 35893811.
Torabi S, Soleimani S, Mahravani H, Ebrahimi MM, Shahsavandi S. Mouse fibroblast L929 cell line as a useful tool for replication and adaptation of infectious bursal disease virus. Arch Razi Inst. 2023 Jun 1;78(3):863-71. doi: 10.22092/ARI.2023.361584.2663, PMID 38028862.
Wang X, Burgess DJ. Drug release from in situ forming implants and advances in release testing. Adv Drug Deliv Rev. 2021 Aug 5;178:113912. doi: 10.1016/j.addr.2021.113912, PMID 34363860.
Mahakul S, Srivastava P, Tiwari S. Atrigel technology: a biodegradable repository for long-term drug release. Tanz J. 2025;20(5):413-24.
The Atrigel drug delivery system. In: Rathbone MJ, Hadgraft J, editors. Modified‑release drug delivery technology. Boca Raton (FL): CRC Press; 2002. p. 671–80. doi: 10.1201/9780203910337-57.
Shobeirean A, Attar H, Varshochian R, Rezvanfar MA. Glatiramer acetate in situ forming gel a new approach for multiple sclerosis treatment. Daru. 2024;32(2):649-64. doi: 10.1007/s40199-024-00532-z, PMID 39225953.
Saha S, Lin X, Zhou L, Xue A, Gosselin E, Chothe PP. Evaluation of the impact of the polymer end groups and molecular weight on in vitro and in vivo performances of PLGA-based in situ forming implants for ketoprofen. J Pharm Sci. 2024 Oct 1;114(1):424-33. doi: 10.1016/j.xphs.2024.10.019, PMID 39426566.
Hu F, Qi J, Lu Y, He H, Wu W. PLGA-based implants for sustained delivery of peptides/proteins: current status challenge and perspectives. Chin Chem Lett. 2023 Feb 24;34(11):108250. doi: 10.1016/j.cclet.2023.108250.
Southard GL, Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert Opin Investig Drugs. 1998 Sep 1;7(9):1483-91. doi: 10.1517/13543784.7.9.1483, PMID 15992045.
Ravivarapu HB, Moyer KL, Dunn RL. Sustained suppression of the pituitary-gonadal axis with an injectable in situ forming implant of leuprolide acetate. J Pharm Sci. 2000;89(6):732-41. doi: 10.1002/(SICI)1520-6017(200006)89:6<732::AID-JPS4>3.0.CO;2-D, PMID 10824131.
Cannella V, Altomare R, Leonardi V, Russotto L, Di Bella S, Mira F. In vitro biocompatibility evaluation of nine dermal fillers on L929 cell line. BioMed Res Int. 2020 May 27;2020:8676343. doi: 10.1155/2020/8676343, PMID 32596390.
Hivechi A, Yousefmoumji H, Bahrami SH, Brouki Milan PB. Fabrication and characterization of in situ gelling oxidized carboxymethyl cellulose/gelatin nanofibers for wound healing applications. Int J Biol Macromol. 2025 Jan 17;298:140033. doi: 10.1016/j.ijbiomac.2025.140033, PMID 39828175.