
Int J App Pharm, Vol 18, Issue 1, 2026, 206-214Original Article
POLYMER STABILIZED AMORPHOUS DISPERSIONS OF CLONAZEPAM: CHARACTERIZATION AND IN VITRO DISSOLUTION ANALYSIS
KHURSHID JAHAN1*, TONMOY BHOWMICK1, JANNATUL FERDOUSI1, SHIHAB HOSSAIN1, NAHID HASAN SHUVO1
Department of Pharmacy, World University of Bangladesh, Uttara, Dhaka-1230, Bangladesh
*Corresponding author: Khurshid Jahan; *Email: jahan2@pharmacy.wub.edu.bd
Received: 06 Aug 2025, Revised and Accepted: 20 Nov 2025
ABSTRACT
Objective: The objective of this study is to enhance the aqueous solubility and dissolution rate of poorly water soluble clonazepam (CLZ) by formulating polymer stabilized amorphous solid dispersions (ASDs) using melting and co-precipitation methods. The study systematically evaluates the effect of various hydrophilic polymers at different drug to polymer ratios on the physicochemical properties, dissolution behavior, and stability of the resulting solid dispersions (SDS). This research aims to optimize polymer selection and preparation techniques to improve the bioavailability and therapeutic efficacy of clonazepam, addressing current gaps in comparative studies of polymer based solubility enhancement strategies for this drug.
Methods: SDS of CLZ were prepared using melting and co-precipitation methods with four hydrophilic polymers (HPMC K4MCR, PEG 6000, Methocel K15, and Kollidon Cl) at drug to polymer ratios of 1:1, 1:3, and 1:5. The formulations were characterized through in vitro dissolution studies, drug content analysis, and structural evaluations using Fourier Transform Infrared Spectroscopy (FTIR), Differential Scanning Calorimetry (DSC), and Scanning Electron Microscopy (SEM).
Results: The pure CLZ showed limited solubility (51±0.577%) at 60 min, whereas polymer ASDs significantly enhanced drug release. The PEG 6000 based formulation (ASD9, 1:5 ratio) exhibited the highest improvement (80.95±0.460%), followed by HPMC K4MCR (ASD3, 75.95±0.033%). Enhanced dissolution is attributed to increased carrier content and inhibition of crystallisation. FTIR and DSC analyses confirmed the absence of chemical interaction, while SEM demonstrated drug amorphisation, supporting improved solubility. ADMET analysis indicated the suitability of clonazepam for solubility enhancement via solid dispersion systems.
Conclusion: Polymer stabilized ASDs improved the aqueous solubility and in vitro dissolution rate of CLZ. Among the polymers tested, PEG 6000 demonstrated the most favourable performance, improving drug release and stability without evidence of chemical interaction. These findings suggest that this approach may hold promise for enhancing the bioavailability and therapeutic efficacy of poorly water-soluble drugs, such as CLZ, although further in vivo studies are needed to confirm these effects.
Keywords: Clonazepam, Binary solid dispersion, Melting method, Release kinetic, FTIR, SEM, DSC
© 2026 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2026v18i1.56438 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Compounds with very low water solubility are often thought to exhibit dissolution rate limited absorption, which results in poor absorption, distribution, and delivery to the target organ. In this situation, increasing water solubility is a worthwhile objective to increase therapeutic efficacy [1]. When the drug is poorly soluble, then the amount of required drug will be inadequate at the target site compared to the amount being administered [2]. The aqueous solubility of a drug in a variety of pH levels is one of its most important properties for oral drug delivery [3]. Therefore, the selection of appropriate polymers is important in the formulation of a new drug that can potentially the solubility of drug molecules [4]. The polymers used to overcome the problem have shown promise by inhibiting crystallisation to ensure a prolonged supersaturated state of the active pharmaceutical ingredient (API) [5]. The oral absorption of almost 75% of the medication is restricted due to its insoluble nature. ASDs is a technology that has shown great promise in increasing the oral bioavailability of insoluble medicines. In the homogeneous ASDs system, amorphous medications are distributed in hydrophilic polymeric carriers at the molecular level. Since the amorphous form has a higher free energy than its crystalline counterpart, it can greatly increase the solubility of intractable medications. One of the most effective methods for enhancing their release is the use of SDS technology [6]. So, the selection of a polymer is the key factor determining the success of ASD development [7]. Nowadays, many new polymers have been studied, being employed widely in SDS technology. Usually, hydrophilic polymers were employed, but recently, hydrophobic swellable polymers have also been preferred as SDS carriers to retard the fast dissolution of the drug for the controlled or sustained release formulations [8]. The polymers used in our research are HPMC (hydroxy propyl methyl cellulose) K4MCR, methocel K15, kollidon Cl and polyethene glycol (PEG) 6000. HPMC is a non-ionic cellulose ether, which is soluble in water, most polar organic solvents such as methanol, ethanol. It is a hydrophilic, biodegradable, and biocompatible polymer having a wide range of applications in drug delivery [9]. Methocel K15 is an HPMC polymer with moderate hydroxypropyl substitution, which is a water soluble cellulose ether polymer. It improves spreadability, rheological properties and facilitates the diffusion of the drug [10]. Kollidon Cl is a crosslinked polymer, a dissolution enhancer to improve the release characteristics of drugs [11]. The polymer PEG is useful to solve the problems of solubility, stability, dissolution and bioavailability. Hence, the relatively low melting points of PEG play an auspicious role in the solubility enhancement of various compounds [12]. PEG is a widely used hydrophilic and nonionic polymer, used to produce steric stabilisation in the formulation that, in turn, reduces the tendency of particles to aggregate, which results in enhanced stability during application and storage of the formulation [13]. We have used commonly used polymers based on their ability to enhance solubility, stabilise the amorphous form of the drug, and improve dissolution profiles.
The Biopharmaceutics Classification System (BCS) classifies drugs according to their water solubility and intestinal permeability [14]. Class II drugs like CLZ have a low solubility in water. It belongs to the class of organic compounds known as 1,4-benzodiazepines, containing a benzene ring fused to a 1,4-azepine, which is practically odorless, freely soluble in methanol, acetone, ethanol, and practically insoluble in water (at 25 °C<0.1 mg/ml) [15].
Fig. 1: 3D structure of clonazepam
The drug is prescribed for the treatment of epilepsies, seizures, and panic disorder. In this situation, increasing the aqueous solubility of CLZ is the objective to get quick therapeutic management with reduced side effects. In some cases, the conventional system possesses several limitations, then SDS formulation can be an alternative, reducing crystallinity and increasing water solubility, thus helping improve therapeutic efficacy.
CLZ directly limits its oral bioavailability and rapid therapeutic response, especially critical in the management of seizures and panic disorders. Although several formulation strategies exist to enhance solubility, few studies have systematically compared multiple hydrophilic polymers across different SDS techniques tailored specifically to CLZ. Furthermore, the existing literature often lacks a comprehensive evaluation combining physicochemical, morphological, and kinetic characterizations. To bridge this gap, the present study employs a dual method (melting and co-precipitation) approach using selected hydrophilic to develop binary SDS. By optimizing polymer type and ratio, this research aims to enhance CLZ solubility, improve dissolution rate, and ultimately, increase its therapeutic potential through better bioavailability and reduced dosing requirements.
MATERIALS AND METHODS
Materials
Pharmaceutical-grade Clonazepam was obtained as a gift sample from Pharmasia Ltd., Bangladesh. The polymers HPMC K4MCR, methocel K15, kollidon Cl and PEG 6000 and the other chemicals like sodium lauryl sulfate (SLS), methanol, ethanol, and distilled water (DW) employed during this research work were analytical grade.
Instruments
Digital Water Bath (XMTD-204, China), Digital balance (PS. P2.610, Taiwan), Vacuum Desiccator (Pyrex 3081-150), Dissolution rate test apparatus (RC100A, Intech, India), UV-VIS Spectrophotometer Double Beam (UV—1900i, Japan), Digital Melting point test apparatus (M3000, Germany), Orbital Shaker (05-3000, USA), Laboratory Drying Oven (DOF-65E, USA), 1951 Microprocessor Tap Density Tester, DSC machine: JSM-7610F (Field Emission Scanning Electron Microscope, Fourier transform Infrared Spectrometer (Model –IR Affinity-IS, MIRacle 10, Shimadzu, Japan were employed for formulation and characterization of the solid dispersion formulations.
Methods
Quantitative solubility analysis
CLZ, the model drug molecule selected for this study, has very poor aqueous solubility. An equilibrium solubility assessment was performed in the presence of various hydrophilic polymers (HPMC K4MCR, Methocel K15, Kollidon Cl and PEG 6000). The physical mixture (PM) of the drug and polymer was added into glass stoppered flasks containing 10 ml of distilled water (DW), phosphate buffer of pH 7.4 and 0.1% sodium lauryl sulfate (SLS) containing DW in the ratios of 1:0 w/w, 1:1 w/w, 1:2 w/w, and 1:3 w/w. The flasks were then sealed properly and shaken on a rotary shaker at room temperature. After equilibration for 96 h, the solutions were filtered through Whatman filter paper No. 41. From the filtrate, 1 ml was withdrawn by syringe and diluted to 100 ml by using the same dissolution media and estimated by UV-Vis spectroscopy at λmax 243 nm [16]. The study was performed in triplicate and calculated as a mean±SD.
Impact of polymers on glass transition temperature (Tg) and drug performance
A polymer with an optimal Tg can maintain the drug in an amorphous, high energy state, facilitating faster dissolution. Polymers with higher Tg can trap the drug molecules within a rigid matrix at room temperature, reducing molecular mobility. This reduces the propensity for the drug to recrystallize back to its thermodynamically favored crystalline state, preserving improved solubility and dissolution.
Preparation of a standard curve of CLZ in distilled water (DW)
To prepare a standard curve, 0.02g of CLZ was accurately weighed and dissolved in 100 ml of methanol to make the solution 0.2 mg/ml. Then 900 ml of DW was added. 1, 2, 3, 4, 5, 6, 7, 8, 9, and 10 ml of the solution were taken in a 10 ml volumetric flask, then DW of different volumes was added, respectively for serial dilution to make different CLZ concentrations. These solutions were then analysed by UV spectrophotometer (UV-VIS Spectrophotometer Double Beam (UV—1900i, Japan) at 243 nm and absorbance was recorded. Finally, the absorbance values were plotted against drug concentration, and a standard curve was produced [17].
Preparation of CLZ SDS and physical mixture (PM)
An accurately weighed amount of drug and polymer in different ratios, as specified in Tables 1 and 2, was systematically mixed for 10 min in a glass mortar. The mixture is then preserved in a screw cap container in a desiccator to be used for inspection [18].
SDS with the melting method consisted of the drug and polymers in varying ratios
The melting method makes the binary complexes of CLZ and selected polymers (proposed by Sekiguchi and Obi in 1961), with the PM of a drug and carrier, then heated directly until it melts. The mixture solidified in an ice bath under vigorous stirring, and the final mass was crushed, pulverized, and sieved. Appropriately, this has undergone many modifications in pouring the homogenous melt in the form of a thin layer onto a stainless steel plate and cooled by cold water on the opposite side of the plate. The technique gives a much finer dispersion of crystallites [19].
SDS with the co-precipitation method consisted of the drug and polymers in varying ratios
In this method, the carrier is first dissolved in a solvent to prepare a solution, and the drug is incorporated into the solution by stirring to form a homogeneous mixture. Then, water is added dropwise to the mixture to induce precipitation. Finally, the precipitate is filtered and dried. The collected sample was kept at room temperature in a screw capped glass vial in a desiccator until use [20].
Analytical characterization of the prepared formulation
Melting Point confirmation
The melting point of CLZ was confirmed by taking a small amount of the sample in a capillary tube closed at one end and placed in a melting point apparatus. The value was noted in triplicate, and the average value was noted to compare with references [21].
Formulation efficiency and API quantification
The batch yield was calculated to evaluate formulation efficiency. An acceptable percent yield (>85%) confirmed good scalability of the technique [22]. However, the API quantification revealed consistent loading across all formulation batches. For these measurements, accurately weighed SDS equivalent to 10 mg of CLZ was transferred to a 10 ml volumetric flask containing 10 ml of DW with 0.2% SLS and the above content was suitably dissolved. 1 ml of this solution was diluted to 10 fold with methanol and analysed by UV-VIS Spectrophotometer Double Beam (UV—1900i, Japan), at 243 nm [23].
The flow properties of CLZ SDS were assessed by determining the bulk density, tapped density, angle of repose, Carr’s index, and Hausner’s ratio.
Table 1: Drug polymer ratio for different formulations by the melting and co-precipitation methods
| Solid dispersions codes | Polymer | Drug: polymer ratio (w/w) | Solid dispersions codes | Polymer | Drug: polymer ratio (w/w) |
| Melting method | Co-precipitation method | ||||
ASD1 ASD2 ASD3 |
HPMC K4MCR | 1:1 1:3 1:5 |
ASD13 ASD14 ASD15 |
HPMC K4MCR | 1:1 1:3 1:5 |
ASD4 ASD5 ASD6 |
Methocel K15 | 1:1 1:3 1:5 |
ASD16 ASD17 ASD18 |
Methocel K15 |
1:1 1:3 1:5 |
ASD7 ASD8 ASD9 |
PEG 6000 | 1:1 1:3 1:5 |
ASD19 ASD20 ASD21 |
PEG 6000 |
1:1 1:3 1:5 |
ASD10 ASD11 ASD12 |
Kollidon Cl | 1:1 1:3 1:5 |
ASD22 ASD23 ASD24 |
Kollidon Cl |
1:1 1:3 1:5 |
Bulk density (BD) and tapped density (TD)
Bulk density (BD) was used to determine the amount of drug that occupies the volume in g/ml. It was then expressed in g/ml and is calculated by using the following formula:
BD =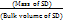
An increase in tapped density suggests improved powder consolidation under mechanical vibration. The tapped volume was measured by tapping the powder 10, 500, and 1250 taps in the TD apparatus (Electro Lab USP II) according to USP and the % variation was calculated from the formula: [24]
TD = 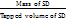
A comparison between the two helps estimate flowability and compressibility.
Compressibility index and Hausner’s ratio
The compressibility index is an important measure that can be obtained from the bulk and tapped densities. According to the theory, the less compressible the material, the more flowable it is [25]. The relationship between the % compressibility indexes with flowability can be given by:
Compressibility index  ×100
×100
The Hausner ratio is usually estimated from the tapped density and bulk density ratio. It is determined from the formula: [26]
Hausner’s ratio = 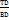
Angle of repose
Lower values of the angle of repose denote superior flow properties. Usually, for most of the pharmaceutical powders, the values range from (25 to 45)°. Angle of repose was calculated by using the formula: [27]
tan θ = 
Ꝋ=tan-1 ()
h = height of heap of pile, r = radius of base of pile
In vitro dissolution studies of pure CLZ and the formulation of SDS with polymers
In vitro drug release studies were performed to evaluate the dissolution behaviour of the pure drug and SDS formulations. A USP Type II (paddle) dissolution apparatus was employed, operating at 75 rpm and maintained at a temperature of 37±0.5 °C. Each formulation was immersed in 900 ml of DW as the dissolution medium. Aliquots were collected at 10-minute intervals, and an equal volume of fresh medium was added to maintain sink conditions. The withdrawn samples were subsequently filtered, appropriately diluted, and analysed spectrophotometrically at a wavelength of 243 nm using a UV–Vis spectrophotometer [28].
Drug release kinetics from the SDS formulation
The dissolution kinetics of different formulations were applied to various kinetic models such as zero order, first order, Higuchi, Hixon-Crowel, and Korsmeyer-Peppas. The value of R2 indicates how well a kinetic model fits the dissolution data. R2 values near 1 denoted the best fitted model. Zero order kinetics characterises a release mechanism in which the relaxation of the polymeric chain controls; here, the release is independent of the polymer concentration. The first order kinetics model suggests that the drug release mechanism is dependent on concentration. The Higuchi model states that the drug release mechanism is square root based on Fick's law and is time dependent. When there is uncertainty about the release mechanism or when there may be more than one release phenomenon at work, the Korsmeyer-Peppas model is employed. It is possible to identify whether the release occurs by anomalous transport, case II transport, Fickian diffusion, or super case II transport based on the values found for the release exponent, n [29]. For a new drug formulation, this model helps predict how the drug will be released over time, allowing for the design of formulations that ensure efficient drug delivery.
Compatibility analysis of drug and polymers using hansen and flory–huggins approaches
The compatibility between CLZ and various polymers (HPMC K4MCR, PEG 6000, Methocel K15, Kollidon Cl) was assessed using both Hansen solubility parameters and Flory–Huggins interaction parameters. The difference in solubility parameters (Δδ) between drug and polymer is calculated as:
Δδ=δtdrug−δtp lymer. ---------------------------------------- (1)
A Δδ value ≤ 7 MPa for Hansen Solubility Parameters (HSP) suggests good miscibility, while values>10 indicate poor compatibility. Further, the Flory Huggins interaction parameter was used to predict thermodynamic miscibility:
χ =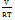 (δt drug−δt p lymer)2 ---------------------------------- (2)
(δt drug−δt p lymer)2 ---------------------------------- (2)
Where:
V = molar volume of the drug (cm³/mol),
R = universal gas constant (8.314 J/mol·K),
T = temperature in Kelvin (typically 298.15 K).
A χ value<0.5 indicates good miscibility, and values>0.5 indicate immiscibility.
The Δδ values for all polymers were within the acceptable miscibility range, suggesting good potential for interaction with CLZ. PEG 6000 showed the smallest Δδ and χ values, indicating the highest miscibility, which correlates with its superior dissolution performance observed in vitro. HPMC K4MCR and Methocel K15 also showed favorable compatibility. Kollidon Cl showed moderate compatibility, which may affect its dispersion efficiency and dissolution enhancement [30, 31].
Characterisation of drug polymer interactions
Molecular interactions between the drug and polymers were explored using spectroscopic and thermal analytical tools. The stability of binary mixtures was assessed to ensure no significant drug polymer incompatibility prior to formulation.
Fourier transform Infrared (FTIR) analysis
Based on dissolution characteristics, an optimised formulation was selected, and a Fourier Transform Infrared (FTIR) was performed to observe drug polymer interaction. The pure CLZ, SDS formulations with PEG 6000 melting method at a 1:5 drug polymer ratio characterised respectively with an FTIR Spectrometer (Model –IR Affinity-IS, MIRacle 10, Shimadzu, Japan). The pure drug and developed SDS formulations were studied employing the KBr disks and scanned in the range of 4000 to 400 cm-1 to investigate the compatibility of the drug with the polymer [32].
Differential scanning calorimetry (DSC)
A heat-flux DSC instrument model 822e (Mettler Toledo, Schwerzenbach, Switzerland) was used to obtain scans of CLZ and its SDS formulation. The technique was widely used for stability studies and measuring the thermal behavior of the prepared SDS [33].
Scanning electron microscopy (SEM)
The drug and selected SDS formulation were analyzed using scanning electron microscopy (SEM) to assess their morphology, surface roughness, fracture, cleavage, and crystal characteristics, which are shown in fig. 7 a) and b) [34].
Statistical analysis
All experimental data were obtained in triplicate and expressed as mean±standard deviation (SD). Statistical analysis was performed to evaluate differences in drug content and dissolution profiles among various solid dispersion formulations (ASD1 to ASD24) and the pure drug. One way ANOVA was employed to determine the significance of variation between formulations, with a confidence level set at p<0.05. The analysis helped identify the formulation exhibiting the most statistically significant improvement in drug release performance. Results are presented as mean±SD (n = 3).
RESULTS AND DISCUSSION
Melting point and solubility
The melting point of CLZ observed at 238 °C, confirms the purity and crystalline nature of the drug. The low solubility of CLZ in DW (0.03±0.00057) mg/ml reflects its poor aqueous solubility, typical for many lipophilic drugs, which often necessitates solubility enhancement strategies for formulation development.
Impact of polymers on (Tg)
The glass transition temperature (Tg) of the SDS was investigated by DSC to understand the effect of polymers on the physical stability of CLZ. Polymers such as PEG 6000, HPMC K4MCR, Methocel K15, and Kollidon Cl increased the Tg of the formulations compared to the pure drug, indicating enhanced molecular rigidity in the polymer matrix. PEG 6000 has a relatively low Tg compared to other polymers used in this study, which promotes greater molecular mobility at physiological temperatures and facilitates the formation of an amorphous dispersion with reduced crystallinity. HPMC, having gel forming capability, controls release but can moderate dissolution rates, while Kollidon Cl, due to its crosslinked nature, limits dissolution enhancement. The dissolution rank order (PEG 6000>HPMC K4MCR>Kollidon Cl>Methocel K15) reflects variations in polymer hydrophilicity, miscibility, and ability to inhibit crystallisation.
Quantitative solubility analysis
The physical mixture (PM) of CLZ and PEG 6000 displayed maximum solubility of (0.03±0.0005) mg/ml compared to the other polymers; however, the solubility of the PM expanded (0.062±0.0513) mg/ml in the presence of 0.1% SLS with DW due to the formation of a micelle (table 2). This result indicated that the PM enhances the aqueous solubility. Since PEG 6000 exhibited a better dissolution rate, only the table for the PM of the drug and PEG 6000 is shown here:
Table 2: Quantitative solubility analysis of the optimised formulation of CLZ and PEG 6000
| PM ratio | Solubility in DW (mg/ml) | Solubility in phosphate buffer PH7.4 (mg/ml) |
Solubility in DW with 0.1% SLS solution (mg/ml) |
| 1:0 | 0.03±0.0005 | 0.042±0.003 | 0.062±0.0513 |
| 1:1 | 0.12±0.0005 | 0.132±0.008 | 0.138±0.0188 |
| 1:2 | 0.16±0.0090 | 0.178±0.0199 | 0.221±0.0251 |
| 1:3 | 0.2±0.0693 | 0.228±0.0356 | 0.249±0.0046 |
PM=Physical mixture, DW=Distilled water, CLZ=Clonazepam. Data is presented as mean±SD (n=3).
The study also states that the solubility of the drug was pH dependent, and it was higher in phosphate buffer (pH 7.4) with (0.042±0.003) mg/ml. This may be the result of weak acidity, which causes it to remain ionized at higher pH values but unionized at lower pH values. On the other hand, the solubility is enhanced in phosphate buffer pH 7.4 due to ionic interactions or other ways. When hydrophilic polymer PEG 6000 was included in the formulation, the solubility improved (0.121±0.0005) mg/ml quickly as the drug got scattered within the polymers, where decreased morphological characteristics led to an increase in bioavailability. When the polymer concentration was amplified 3 times, the solubility was boosted concurrently (0.16±0.0090) mg/ml due to better scattering into the polymeric framework, which encouraged better solubility in DW and (0.178±0.0199) mg/ml in phosphate buffer (pH 7.4). Additionally, the solubility of the PM with the highest polymer ratio was observed in the presence of 0.1% SLS with DW (0.249±0.0046) mg/ml. Similarly, improved solubility was observed with 1:5 ratio of drug and PEG 6000, that is (0.228±0.0356) mg/ml in buffer solution and (0.249±0.0046) mg/ml at 0.1% SLS solution.
The enhanced dissolution and solubility observed in the SDS formulations of CLZ in this study align well with existing literature that demonstrates polymer stabilized amorphous dispersion ability to overcome poor aqueous solubility of BCS class II drugs. The significant improvement with PEG 6000 based formulation, with the melting method, reported PEG 6000 is effective in enhancing gliclazide dissolution through SDS due to its low melting point and steric stabilization properties.
Compatibility analysis of drug and polymers using hansen and flory–huggins approaches
HSP values were derived for both the drug and the polymers via the group contribution method. The distance (Ra) between CLZ and PEG 6000 was found to be below the miscibility threshold, suggesting good compatibility. Similarly, Flory Huggins (χ) calculations, using the obtained HSP values and DSC derived melting point depression, consistently yielded values below 0.5 for CLZ PEG 6000, denoting a thermodynamically favourable interaction. These findings corroborate the absence of new peaks in FTIR and the partial amorphization observed in DSC, collectively confirming the physical and thermodynamic compatibility of CLZ and PEG 6000 in the SDS.
Micromeritic profiling and process efficiency of binary SDS
The formulation efficiency of the PM of CLZ with various polymers ranged from 85.71±0.060% to 89.41±0.055%, indicating efficient formulation processes (table 3). Key physicochemical factors influencing solubility include particle size reduction, increased porosity, and improved wettability. The incorporation of hydrophilic polymers promotes intimate mixing at the molecular level, allowing the drug to occupy intermolecular spaces within the polymer chains. This close proximity enhances the flexibility of the polymer matrix and facilitates better drug dispersion. Furthermore, the micromeritic evaluations confirmed excellent compressibility and flowability of the SDS powders, supporting their suitability for downstream processing in oral solid dosage forms.
Table 3: Pharmaceutical characteristics of PM of CLZ with different polymers
| Formulation | Formulation efficiency (%±SD) | Bulk density (%±SD) | Tapped density (%±SD) | Compressibility index (%±SD) | Hausner ratio (%±SD) | Angle of repose (%±SD) |
Flowability |
| CLZ: HPMC K4MCR | 88.23±0.023 | 0.28±0.005 | 0.32±0.007 | 13.19±0.020 | 1.15±0.005 | 26.56±0.058 | Excellent |
| CLZ: Methocel K15 | 89.34±0.035 | 0.56±0.01 | 0.62±0.005 | 9.56±0.047 | 1.1±0.026 | 25.55±0.080 | Excellent |
| CLZ: Kollidon Cl | 85.71±0.060 | 0.41±0.044 | 0.47±0.005 | 12.76±0.384 | 1.15±0.08 | 26.27±0.060 | Excellent |
| CLZ: PEG 6000 | 89.41±0.055 | 0.41±0.005 | 0.46±0.005 | 10.25±0.060 | 1.15±0.04 | 26.27±0.005 | Excellent |
Data is presented as mean±SD (n=3).
The formulation efficiency was found to be consistently high, ranging between (85.71±0.060)% and (89.41±0.055)%, indicating successful incorporation of the drug into the polymeric matrix. Among the polymers evaluated, PEG 6000 demonstrated superior drug dispersion within the matrix compared to the others used in the SDS formulations. The efficiency values suggest minimal drug loss during formulation, with drug content measured at (94±0.577)%, which lies well within acceptable pharmacopeial limits. This highlights that the developed formulations achieved efficient drug loading and uniform distribution, ensuring reproducibility and quality of the final product.
Effects of different concentrations of polymer on in vitro drug release
The SDS formulations improve the dissolution rate compared to the pure drug. The study was conducted in a calibrated six basket dissolution test apparatus with DW as a dissolution (900 ml) maintained at (37 ± 0.5) °C and 75 rpm. About 24 formulations were made. 10 ml of each sample was withdrawn in 10, 20, 30, 40, 50 and 60 minute intervals and replaced with identical volumes of the fresh dissolution media to maintain the sink condition. The collected samples were filtered using a syringe filter (having 0.45 μm pore size), suitably diluted with DW and assayed spectrophotometrically at 243 nm. In vitro dissolution tests were carried out in triplicate (n = 3±SD).
The dissolution enhancement of different polymers is significantly shown in (fig. 4 and fig. 5). The polymer methocel K15 for the melting method and kollidon Cl for the co-precipitation method showed the least dissolution rate. The reasons may be inadequate dispersion of the drug within the polymer matrix, which can lead to inappropriate interaction of the drug and the polymer. The dissolution improvement properties were significant for different polymers. In every case, the highest carrier containing formulation showed a greater dissolution rate (p<0.05). This is because the use of different carriers generally changes the nature of the insoluble drug to an amorphous form that helps to enhance the rate of dissolution. The other mechanism of increasing dissolution is the reduction of crystal size and the physical-chemical properties of the polymers used in SDS formulations. The carrier's HPMC K4MCR in the melting method showed maximum drug release at 60 min with ASD3 formulation, which is (75.95±0.03)% and PEG 6000 with ASD9 formulation at 60 min (80.95±0.46)% drug release, respectively. Furthermore, SDS formulation with the co-precipitation method with PEG 6000 showed (69.85±0.47)% drug release at 60 min, while pure drug showed (51±0.57)% release. However, the dissolution rate rises in the order PEG 6000>HPMC K4MCR>Kollidon Cl>Methocel K15 for the melting method, in addition to PEG 6000>Methocel K15>HPMC K4MCR>Kollidon Cl for the co-precipitation method. So, the dissolution rate with PEG 6000 significantly enhances drug dissolution (p<0.05) compared to the other polymers used in our current research. Additionally, PEG 6000 is the best polymer amongst the polymers used during the studies. The finer, more homogeneous dispersions with lower residual crystallinity of the melting method boost drug release compared to co-precipitation, accounting for the observed superior performance of PEG 6000 in melting method solid dispersions.
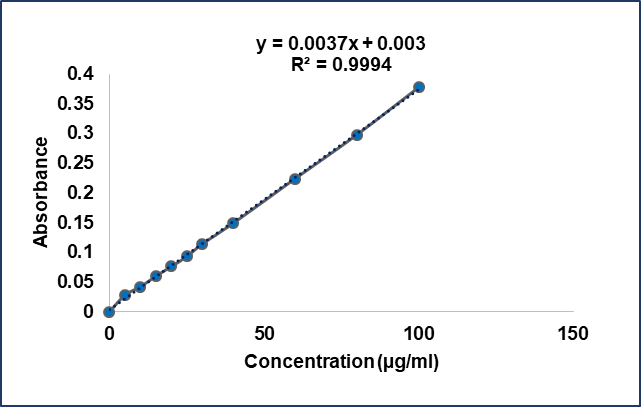
Fig. 2: Calibration curve of CLZ
Kinetics modelling of drug release
The drug release mechanism and kinetics of the drug from the SDS are analysed using zero order, first order kinetics, Higuchi model, and Korsmeyer-Peppas kinetic model. In our study, the first-order and Higuchi diffusion models produced higher R² values compared to the zero-order model (table 4). Therefore, except for ASD1, ASD2, ASD5, ASD11, ASD13, ASD14, ASD15, and ASD23, most of the formulations adhered to the first-order model. Almost all formulations, except ASD1, followed the Higuchi diffusion model. The prevalence of the first order model could be explained by the hydrophilic polymers dissolving more quickly, creating a boundary layer that enables drug particles to dissolve thoroughly. Additionally, this model explains how the quantity of hydrophilic polymers in the prepared systems influences the drug dissolution rate from the created SDS. These findings closely align with the conclusions drawn from the measured dissolution parameters. Release kinetic study revealed that maximum formulations (ASD3, ASD4, ASD6, ASD8, ASD9 and ASD10, ASD12, ASD16, ASD17, ASD19, ASD22, ASD23) displayed zero order release model and except for ASD1, ASD2, ASD5, ASD11, ASD13, ASD14, ASD15, ASD23, every formulation displayed 1st order release pattern. Moreover, except for ASD1, almost every formulation exhibits the Higuchi release model and some formulations ASD2, ASD4, ASD7, ASD10, ASD13, ASD13 and ASD15-ASD24, exhibit both the Higuchi and Korsmeyer-Peppas model of drug release. The predominance of first-order and Higuchi models across formulations indicates that drug release is largely concentration dependent and governed by diffusion through a polymer matrix or boundary layer, consistent with hydrophilic polymer systems. Most formulations showed super case II transport with a release exponent. n>0.89, suggesting that drug release is primarily controlled by polymer relaxation and erosion mechanisms rather than simple diffusion. Together, these findings highlight that both polymer properties and drug–polymer interactions determine dissolution enhancement and underscore the importance of selecting polymers that balance stability with rapid release. Understanding these mechanisms helps optimize polymer selection and formulation techniques to achieve the desired release profile, guiding dosage form design to improve therapeutic outcomes for CLZ.
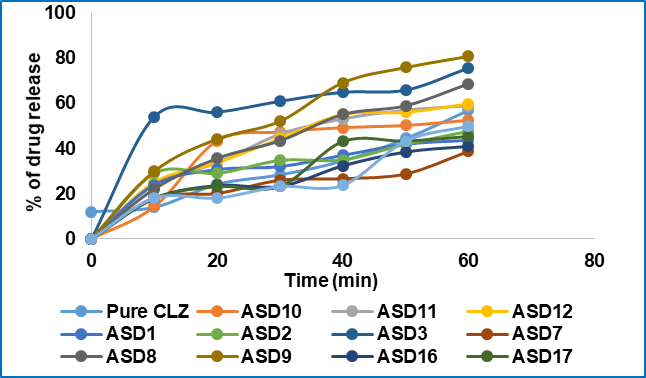
Fig. 3: Comparative dissolution studies of pure CLZ and prepared SDS from the melting method. Condition: distilled water; RPM: 75; 37 C±0.5 °C
Table 4: Correlation coefficient (R2) values for the SDS formulation of different methods
| Formulation code | Correlation coefficient (R2) value | ||||||
| Zero order release plot | First order release plot | Higuchi release plot | Hixon crowel release plot | Korsmeyer peppas plot | n | SDS techniques | |
| Pure CLZ | 0.990 | 0.979 | 0.910 | 0.780 | 0.995 | 0.957 | MM |
| ASD1 | 0.864 | 0.823 | 0.760 | 0.717 | 0.394 | 0.609 | |
| ASD2 | 0.756 | 0.836 | 0.952 | 0.523 | 0.980 | 0.602 | |
| ASD3 | 0.919 | 0.963 | 0.960 | 0.642 | 0.528 | 0.935 | |
| ASD4 | 0.900 | 0.932 | 0.980 | 0.650 | 0.919 | 0.609 | |
| ASD5 | 0.846 | 0.891 | 0.983 | 0.565 | 0.841 | 0.602 | |
| ASD6 | 0.903 | 0.958 | 0.993 | 0.605 | 0.823 | 1.005 | |
| ASD7 | 0.830 | 0.960 | 0.973 | 0.574 | 0.936 | 0.873 | |
| ASD8 | 0.951 | 0.981 | 0.962 | 0.682 | 0.653 | 0.586 | |
| ASD9 | 0.921 | 0.944 | 0.943 | 0.645 | 0.527 | 0.983 | |
| ASD10 | 0.939 | 0.956 | 0.944 | 0.694 | 0.911 | 0.804 | |
| ASD11 | 0.796 | 0.824 | 0.970 | 0.539 | 0.982 | 0.586 | |
| ASD12 | 0.908 | 0.942 | 0.968 | 0.631 | 0.659 | 0.983 | |
| ASD13 | 0.804 | 0.827 | 0.948 | 0.511 | 0.918 | 1.007 | Co-PPT |
| ASD14 | 0.666 | 0.729 | 0.893 | 0.476 | 0.932 | 0.005 | |
| ASD15 | 0.885 | 0.885 | 0.973 | 0.597 | 0.932 | 0.999 | |
| ASD16 | 0.936 | 0.960 | 0.976 | 0.641 | 0.956 | 0.915 | |
| ASD17 | 0.917 | 0.937 | 0.930 | 0.641 | 0.934 | 0.993 | |
| ASD18 | 0.099 | 0.968 | 0.994 | 0.434 | 0.937 | 1.052 | |
| ASD19 | 0.945 | 0.962 | 0.970 | 0.655 | 0.962 | 0.992 | |
| ASD20 | 0.893 | 0.913 | 0.936 | 0.609 | 0.946 | 0.927 | |
| ASD21 | 0.893 | 0.945 | 0.983 | 0.601 | 0.939 | 0.799 | |
| ASD22 | 0.973 | 0.973 | 0.925 | 0.722 | 0.979 | 0.949 | |
| ASD23 | 0.996 | 0.860 | 0.970 | 0.539 | 0.981 | 1.039 | |
| ASD24 | 0.073 | 0.926 | 0.976 | 0.591 | 0.983 | 1.073 | |
(ASD1-ASD3) and (ASD13-ASD15)= formulation with HPMC K4MCR, (ASD4-ASD6) and (ASD16-ASD18) = formulation with methocel K15, (ASD7-ASD9) and (ASD19-ASD21) = formulation with PEG 6000; (ASD10-ASD12) and (ASD22-ASD24) = formulation with kollidon Cl. MM = melting method, Co-PPT = co-precipitation method
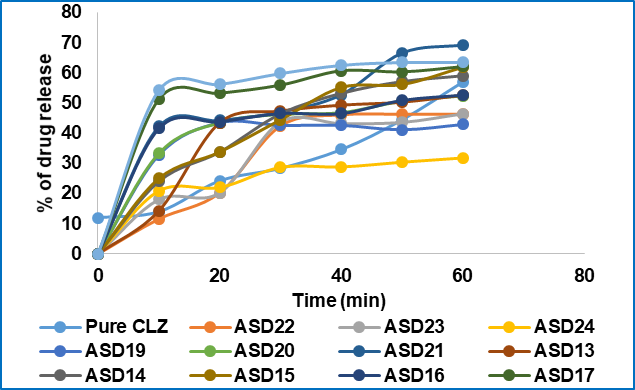
Fig. 4: Comparative dissolution studies of pure CLZ and prepared SDS from the co-precipitation method. Condition: distilled water; RPM: 75; 37 °C±0.5 °C
Fourier transform infrared spectroscopy (FTIR) analysis
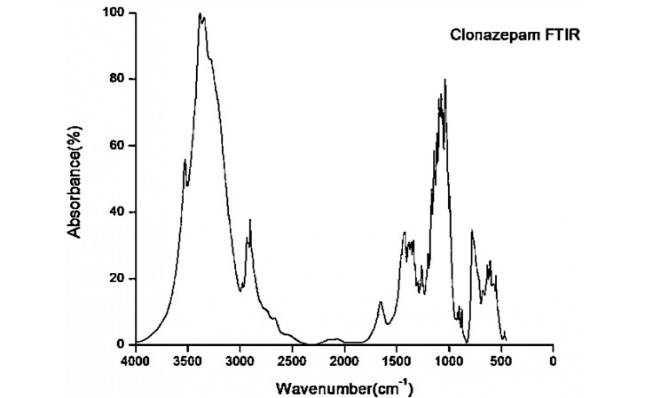
For conducting a compatibility study of the drug and optimum polymer, samples were scanned in the range of 4000 cm-1 to 400 cm-1. FT-IR spectra of the pure CLZ showed prominent peaks at 3500 cm-1, 2100 cm-1, 1563 cm-1, 1228 cm-1, 1651 cm-1,500 cm-1, 600 cm-1, 800 cm-1, respectively (fig. 6).
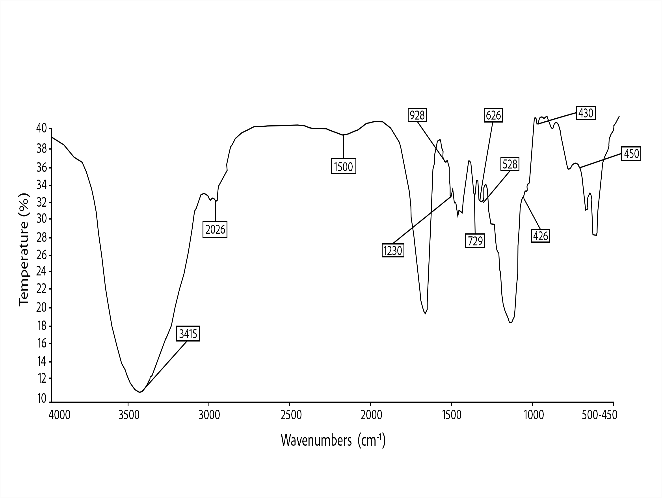
The characteristic peaks of CLZ were largely retained in the SDS formulation with PEG 6000, though some shift and reduction in intensity were noted. The absence of new peaks and the retention of fundamental functional groups indicate no chemical interaction between CLZ and PEG 6000. The peak changes suggest close physical mixing and possible hydrogen bonding, but do not indicate incompatibility. For the best formulation (ASD9, CLZ: PEG 6000, 1:5 ratio), FTIR confirmed preservation of drug integrity within the polymer matrix.
Differential scanning calorimetry (DSC)
The thermograph (DSC) of pure CLZ and SDS formulation with optimum formulation with PEG 6000 was evaluated for drug-polymer compatibility, and the results are shown in fig. 6.
a) b)
Fig. 5: Fourier transform infrared spectrograms of a) Clonazepam, b) Optimised formulation of CLZ with PEG 6000 at a 1:5 ratio in the melting method
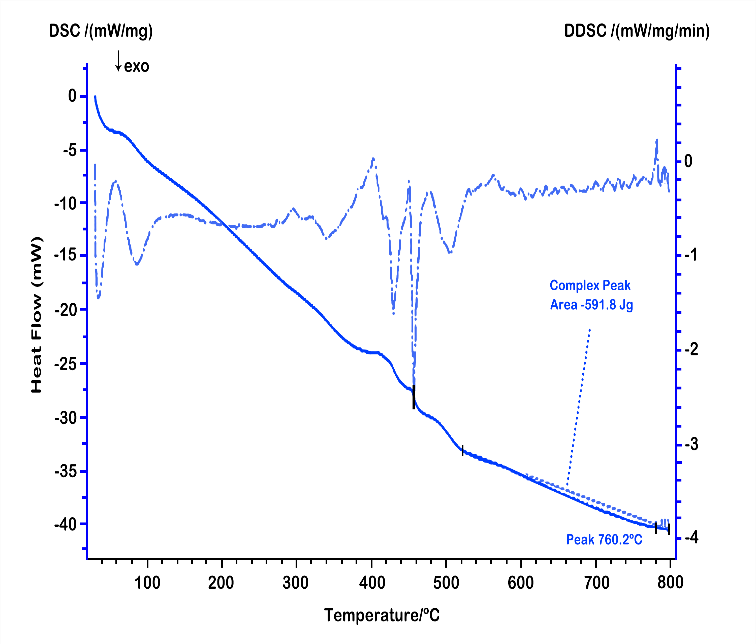
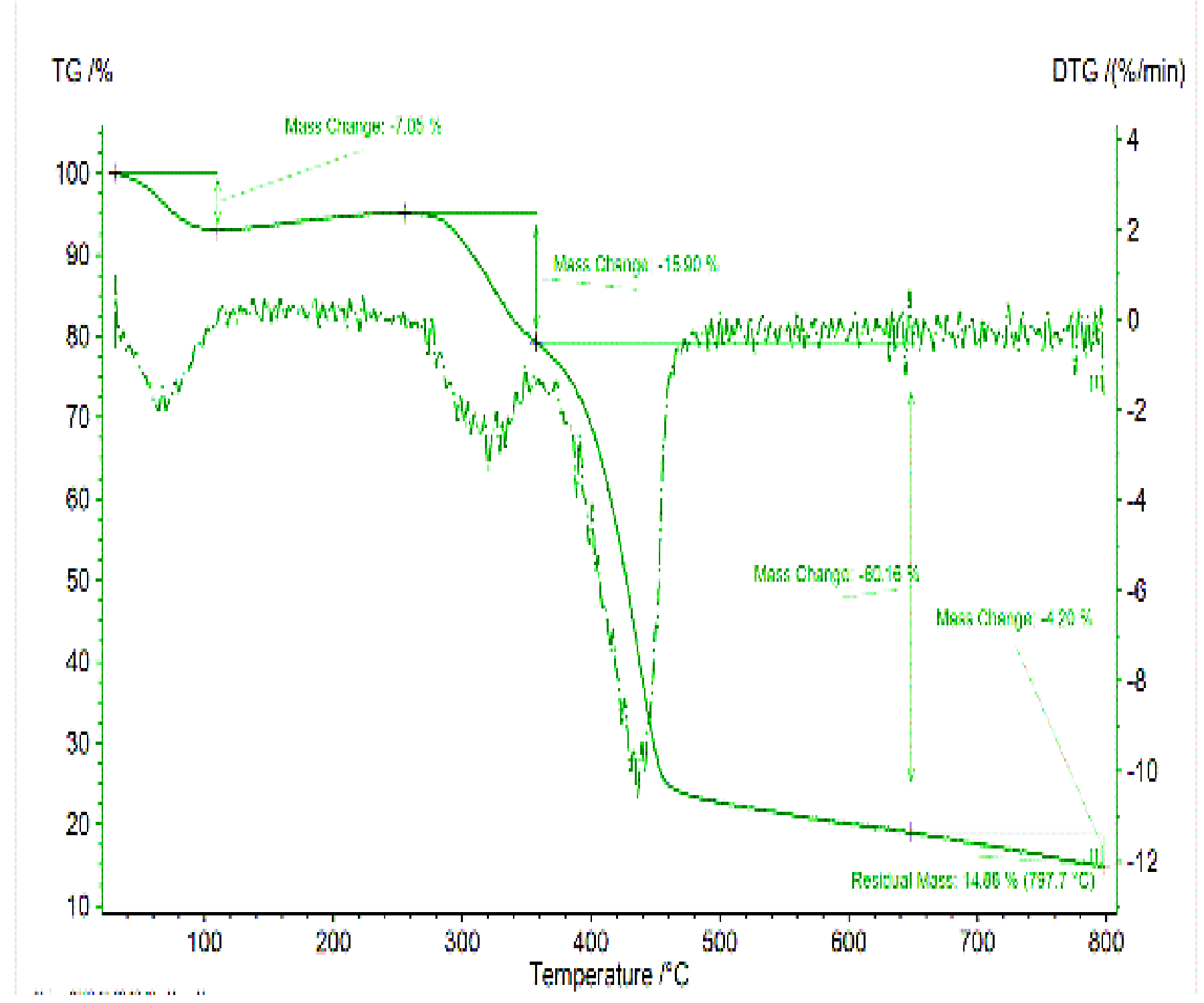
Fig. 6: DSC diagram of the optimised formulation of CLZ SDS in the melting method
The pure CLZ exhibits a sharp endothermic peak at approximately 239 °C, corresponding to its melting point, which confirms its crystalline nature. In contrast, the SDS formulation shows either a reduced intensity or complete disappearance of this endothermic peak, indicating the transformation of CLZ from its crystalline form to an amorphous state when dispersed in the polymer matrix. The polymer (e. g., PEG 6000) typically displays a broad melting peak around 60–65 °C, which may still be observable in the SDS thermogram depending on the drug polymer ratio and miscibility. The absence or reduction of the CLZ melting endotherm in the SDS confirms successful molecular dispersion and supports the improved solubility and dissolution rate observed in subsequent in vitro studies. This thermal behaviour also suggests good compatibility between the drug and polymer, with no evidence of degradation or undesirable interactions during processing.
Scanning electron microscopy (SEM)
The scanning electron micrograph (SEM) of CLZ revealed that pure CLZ particles appeared as irregular shapes, crystalline solids with rough surfaces.

Fig. 7: a) SEM image of pure clonazepam and b) the optimised formulation of clonazepam with PEG 6000 (Melting Method, ASD9)
The SEM exhibited that the optimized SDS formulation with PEG 6000 showed particles with smooth surfaces. The micrograph exhibited a different arrangement between the drug and polymer, indicating incorporation of CLZ in the polymer. In the SDS system, the drug homogeneously disperses into the polymer. The porosity, which is observed in the SDS micrograph, promotes water penetration into the powder and, as such, dissolution of the drug from the SDS system gets enhanced. SEM analysis was performed to compare the morphological differences between the SDS and the physical mixture of CLZ with PEG 6000. Fig. 7. b) shows the surface morphology of the SDS, prepared by the melting method. The image displays a smooth, homogenous surface, suggesting successful incorporation of CLZ into the PEG 6000 matrix in an amorphous or molecularly dispersed form. In contrast, fig. 7. a) represents the physical mixture, where discrete crystalline structures are clearly visible. The rough and heterogeneous morphology indicates that the drug remains in its crystalline state, with no significant interaction or dispersion in the polymer matrix.
ADMET (Absorption, distribution, metabolism, excretion, and toxicity) analysis and drug likeness prediction
ADMET analysis is a vital component of drug discovery and development in computer aided drug designing, as it helps to predict how a drug behaves in the body, how well it can be absorbed, distributed, metabolized, how it is excreted, and whether it could cause any toxic effects [35]. Computational prediction of ADMET properties was performed using the Swiss ADME online platform, which applies multiple rules based and predictive algorithms, including Lipinski’s Rule of Five, Veber’s rule, and the BOILED Egg model for absorption and brain penetration. The canonical SMILES of CLZ was used as input. The analysis confirmed compliance with Lipinski’s rule (molecular weight 315.7 Da, LogP 2.5, H bond acceptors = 4, H bond donors = 1), suggesting good oral bioavailability potential. The topological polar surface area (TPSA) was calculated as 84.6 Ų, within the range typically associated with favourable absorption (<140 Ų). The BOILED-Egg model predicted high passive gastrointestinal absorption but low probability of blood brain barrier penetration, consistent with the known profile of the drug.
The development of polymer stabilised ASDs of CLZ significantly influenced its ADMET properties, particularly absorption. By converting CLZ into an amorphous state and dispersing it within hydrophilic carriers such as PEG 6000 and HPMC K4MCR, solubility and dissolution were markedly improved, leading to enhanced oral absorption and bioavailability. The higher plasma exposure resulting from improved absorption is also expected to promote more efficient tissue distribution. Therefore, the modification of CLZ into amorphous dispersions improved its absorption and distribution profile, without introducing unfavourable changes in metabolism, excretion, or safety. Overall, computational assessments support the drug likeness of CLZ and highlight the potential benefit of ASDs in improving oral absorption.
CONCLUSION
This study demonstrated that SDS prepared with various hydrophilic polymers significantly enhanced the dissolution and solubility of CLZ. ADMET analysis confirmed the favorable pharmacokinetic and toxicity profile of the drug, supporting its potential for novel formulation development. Among polymers, PEG 6000 based SDS showed the greatest improvement in solubility compared to pure CLZ, as supported by DSC and SEM data indicating a crystalline to amorphous transition. The melting method outperformed co-precipitation, likely due to more uniform drug polymer dispersion and finer particles, resulting in faster dissolution. These findings confirm that SDS technology effectively improves the bioavailability of poorly soluble drugs like CLZ, which may reduce dosage requirements and minimize toxicity. Further in vivo studies are needed to validate these results.
ACKNOWLEDGMENT
The authors are thankful to Pharmasia Ltd, Dhaka, Bangladesh, for their generous gift of Clonazepam. The authors are also thankful to Advanced Research Laboratories, Department of Pharmacy, World University of Bangladesh, and the Bangladesh Council of Scientific and Industrial Research (BCSIR) for their support and cooperation in conducting the research.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
JK: Conceptualization and design of the research framework, oversight and supervision of the study execution, data analysis, and preparation of the manuscript. BT: Draft writing of the manuscript, data interpretation, and conducted research in the Laboratory. HS, SHN and FJ: conducted research in the Laboratory.
CONFLICS OF INTERESTS
The authors declare no conflict of interest
REFERENCES
Patel R, Purohit N. Physico-chemical characterization and in vitro dissolution assessment of clonazepam-cyclodextrins inclusion compounds. AAPS PharmSciTech. 2009;10(4):1301-12. doi: 10.1208/s12249-009-9321-3, PMID 19885735.
Rahman A, Haider MF. Solubility of drugs their enhancement factors affecting and their limitations: a review. Int J Pharm Sci Rev Res. 2023;79(2):78-94. doi: 10.47583/ijpsrr.2023.v79i02.014.
Loh ZH, Samanta AK, Sia Heng PW. Overview of milling techniques for improving the solubility of poorly water-soluble drugs. Asian J Pharm Sci. 2015;10(4):255-74. doi: 10.1016/j.ajps.2014.12.006.
Choi MJ, Woo MR, Choi HG, Jin SG. Effects of polymers on the drug solubility and dissolution enhancement of poorly water-soluble rivaroxaban. Int J Mol Sci. 2022;23(16):9491. doi: 10.3390/ijms23169491, PMID 36012748.
Frank DS, Matzger AJ. Probing the interplay between amorphous solid dispersion stability and polymer functionality. Mol Pharm. 2018;15(7):2714-20. doi: 10.1021/acs.molpharmaceut.8b00219, PMID 29924614.
Zhao P, Han W, Shu Y, Li M, Sun Y, Sui X. Liquid–liquid phase separation drug aggregate: merit for oral delivery of amorphous solid dispersions. J Control Release. 2023;353:42-50. doi: 10.1016/j.jconrel.2022.11.033, PMID 36414193.
Zhang J, Guo M, Luo M, Cai T. Advances in the development of amorphous solid dispersions: the role of polymeric carriers. Asian J Pharm Sci. 2023;18(4):100834. doi: 10.1016/j.ajps.2023.100834, PMID 37635801.
Patel K, Shah S, Patel J. Solid dispersion technology as a formulation strategy for the fabrication of modified release dosage forms: a comprehensive review. Daru. 2022;30(1):165-89. doi: 10.1007/s40199-022-00440-0, PMID 35437630.
Deshmukh K, Basheer Ahamed M, Deshmukh RR, Khadheer Pasha SK, Bhagat PR, Chidambaram K. Biopolymer composites with high dielectric performance: interface engineering. In: Biopolymer composites in electronics. Amsterdam: Elsevier; 2017. p. 27-128. doi: 10.1016/B978-0-12-809261-3.00003-6.
Chaurasiya AK, Ansary J, Rahaman MS, Alam ME. Preparation and in vitro evaluation of lamivudine matrix tablets for oral sustained release drug delivery system using Methocel K15M CR polymer. Int J Pharm Sci Res. 2015;6(1):164. doi: 10.13040/IJPSR.0975-8232.6(1).164-71.
Jagtap PS, Tagad RR, Shendge RS. A brief review on Kollidon. J Drug Deliv Ther. 2019;9(2):493-500. doi: 10.22270/jddt.v9i2.2539.
Biswal S, Sahoo J, Murthy PN, Giradkar RP, Avari JG. Enhancement of dissolution rate of gliclazide using solid dispersions with polyethylene glycol 6000. AAPS PharmSciTech. 2008;9(2):563-70. doi: 10.1208/s12249-008-9079-z, PMID 18459056.
Kar M, Chourasiya Y, Maheshwari R, Tekade RK. Current developments in excipient science. In: Basic fundamentals of drug delivery. Amsterdam: Elsevier; 2019. p. 29-83. doi: 10.1016/B978-0-12-817909-3.00002-9.
Malkawi R, Malkawi WI, Al Mahmoud Y, Tawalbeh J. Current trends on solid dispersions: past present and future. Adv Pharmacol Pharm Sci. 2022;2022:5916013. doi: 10.1155/2022/5916013, PMID 36317015.
Soltanpour S, Bastami Z, Sadeghilar S, Kouhestani M, Pouya F, Jouyban A. Solubility of clonazepam and diazepam in polyethylene glycol 200, propylene glycol N-methyl pyrrolidone ethanol and water at (298.2 to 318.2) K and in binary and ternary mixtures of polyethylene glycol 200, propylene glycol, and water at 298.2 K. J Chem Eng Data. 2013;58(2):307-14. doi: 10.1021/je3009842.
Dangre PV, Godbole MD, Ingale PV, Mahapatra DK. Improved dissolution and bioavailability of eprosartan mesylate formulated as solid dispersions using conventional methods. Indian J Pharm Educ Res. 2016;50(3s):S209-17. doi: 10.5530/ijper.50.3.31.
Azad AK, Jahan K, Sathi TS, Sultana R, Abbas SA, UddinUddin AB. Improvement of dissolution properties of albendazole from different methods of solid dispersion. J Drug Deliv Ther. 2018;8(5):475-80. doi: 10.22270/jddt.v8i5.1942.
Jahan K, Akter M, Bhowmick T, Md Harun OR Rashid, Tasnim S, Rahman Rimi R. Effects of various polymers on dissolution improvement of fabricated amorphous clonazepam solid dispersion-an in vitro study. Malays J Pharm Sci. 2024;22(2):81-95. doi: 10.21315/mjps2024.22.2.6.
Mayersohn M, Gibaldi M. New method of solid-state dispersion for increasing dissolution rates. J Pharm Sci. 1966;55(11):1323-4. doi: 10.1002/jps.2600551138, PMID 5969799.
Sonali D, Tejal S, Vaishali T, Tejal G. Silymarin-solid dispersions: characterization and influence of preparation methods on dissolution. Acta Pharm. 2010;60(4):427-43. doi: 10.2478/v10007-010-0038-3, PMID 21169135.
Sisodiya M, Saudagar R. Solubility enhancement formulation development and evaluation of immediate release tablet of antihypertensive drug tadalafil. J Drug Delivery Ther. 2018;8(5):294-302. doi: 10.22270/jddt.v8i5.1872.
Sumana N, Chhitij T. Formulation and enhancement of dissolution rate of poorly aqueous soluble drug aceclofenac by solid dispersion method: in vitro study. Afr J Pharm Pharmacol. 2020;14(1):1-8. doi: 10.5897/AJPP2019.5104.
Mohana M, Vijayalakshmi S. Development and characterization of solid dispersion-based orodispersible tablets of cilnidipine. Beni-Suef Univ J Basic Appl Sci. 2022;11(1):83. doi: 10.1186/s43088-022-00259-3.
Gangane PS, Ghughuskar SH, Mahapatra DK, Mahajan NM. Evaluating the role of Celosia argentea powder and fenugreek seed mucilage as natural super-disintegrating agents in gliclazide fast disintegrating tablets. Int J Curr Res Rev. 2020;12(17):101-8. doi: 10.31782/IJCRR.2020.12173.
Ali IS, Sajad UA, Abdul Rasool BK. Solid dispersion systems for enhanced dissolution of poorly water-soluble candesartan cilexetil: in vitro evaluation and simulated pharmacokinetics studies. PLOS One. 2024 Jun 6;19(6):e0303900. doi: 10.1371/journal.pone.0303900, PMID 38843120.
Gangane PS, Mule VM, Mahapatra DK, Mahajan NM, Sawarkar HS. Development of fenofibrate solid dispersions for the plausible aqueous solubility augmentation of this BCS class-II drug. Int J Curr Res Rev. 2021;13(10):107-16. doi: 10.31782/IJCRR.2021.131006.
Upadhyay S, Shende R, Parmar T, Gupta C. Optimization of metoclopramide fast-dissolving tablets: formulation development and evaluation. Panacea J Pharm Pharm Sci. 2023;12(4):12-21.
Bolourchian N, Mahboobian MM, Dadashzadeh S. The effect of PEG molecular weights on dissolution behavior of simvastatin in solid dispersions. Iran J Pharm Res. 2013;12(Suppl):11-20. PMID 24250667.
Sanchez Aguinagalde O, Sanchez Rexach E, Polo Y, Larranaga A, Lejardi A, Meaurio E. Physicochemical characterization and in vitro activity of poly(ε-caprolactone)/mycophenolic acid amorphous solid dispersions. Polymers. 2024;16(8):1088. doi: 10.3390/polym16081088, PMID 38675007.
Vijayalakshmi S, Subramanian S, Malathi S. Hansen solubility parameter approach in the screening of lipid excipients for the development of lipid nano carriers. Indian J Pharm Educ Res. 2025;59(1s):s71-80. doi: 10.5530/ijper.20254852.
Klueppelberg J, Handge UA, Thommes M, Winck J. Composition dependency of the flory–huggins interaction parameter in drug–polymer phase behavior. Pharmaceutics. 2023;15(12):2650. doi: 10.3390/pharmaceutics15122650, PMID 38139992.
Dhawale P, Mahajan NM, Mahapatra DK, Mahajan UN, Gangane PS. K15M and Carbopol 940 mediated fabrication of ondansetron hydrochloride intranasal mucoadhesive microspheres. J Appl Pharm Sci. 2018 Aug 31;8(8):75-83. doi: 10.7324/JAPS.2018.8812.
Gill P, Moghadam TT, Ranjbar B. Differential scanning calorimetry techniques: applications in biology and nanoscience. J Biomol Tech. 2010;21(4):167-93. PMID 21119929.
Fitriani L, Afriyanti I, Afriyani A, Ismed F, Zaini E. Solid Dispersion of usnic acid-HPMC 2910 prepared by spray drying and freeze drying techniques. Orient J Chem. 2018;34(4):2083-8. doi: 10.13005/ojc/3404048.
Debnath P, Roy UK, Zaman F, Mukherjee PK, Kard A. Exploring the cucurbitacin E (CuE) as an anti-lung cancer lead compound through molecular docking, ADMET, pass prediction and drug likeness analysis. Trop J Nat Prod Res. 2024;8(2):6250-60. doi: 10.26538/tjnpr/v8i2.24.