
Int J App Pharm, Vol 18, Issue 2, 2026, 272-281Original Article
PREPARATION AND CHARACTERIZATION OF SOLID DISPERSION LOADED WITH SELEXIPAG AS MODEL DRUG
DOAA HUSSEIN KARAM1*, MAZIN THAMIR ABDUL-HASAN2
1Babil Health Directorate, Babil, Hillah, Babylon Governorate, Iraq. 2Department of Pharmaceutics, Faculty of Pharmacy University of Kufa, Najaf, Iraq
*Corresponding author: Doaa Hussein Karam; *Email: doaahk83@gmail.com
Received: 13 Sep 2025, Revised and Accepted: 22 Dec 2025
ABSTRACT
Objective: The study aims to prepare solid dispersions of selexipag to enhance its aqueous solubility using two different techniques—solvent evaporation and kneading.
Methods: Twenty-seven solid dispersion (SD) formulations of selexipag were prepared using two different techniques, solvent evaporation (SE) and kneading (KN). Various hydrophilic carriers were employed, including urea, polyethylene glycol 4000 (PEG 4000), polyethylene glycol 6000 (PEG 6000), poloxamer 188 (PXM 188), and poloxamer 407 (PXM 407), at drug-to-polymer ratios of 1:1, 1:3, and 1:5. The prepared formulations were evaluated for saturation solubility, drug content, percentage yield, and in vitro dissolution. Fourier transform infrared spectroscopy (FTIR) was performed to assess drug-polymer compatibility.
Results: The saturation solubility was enhanced by all prepared solid dispersion formulations. The pure selexipag exhibited a solubility of 1.5±0.04 µg/ml, whereas the optimized formulation (F2, PXM407:Selexipag 3:1) prepared by the solvent evaporation method showed a solubility of 25.9±0.2 µg/ml, representing approximately a 17-fold increase compared to the pure drug. The optimized formulation also demonstrated a faster dissolution rate, with 88% of the drug released within 30 min. FTIR results further confirmed drug-carrier compatibility; no additional peaks were observed in any of the binary systems, indicating the absence of chemical interaction between selexipag and the polymer.
Conclusion: Solid dispersion, particularly using poloxamer 407 via solvent evaporation, proved to be an effective and promising approach for enhancing the solubility, dissolution rate, and overall biopharmaceutical performance of selexipag.
Keywords: Dissolution rate, Polaxamer 407, Polymer, Selexipag, Solid dispersion, Solubility, Solvent evaporation
© 2026 The Authors.Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2026v18i2.56830 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
In pharmaceutical development, poorly water-soluble drugs are a common occurrence and represent one of the most significant challenges [1]. Approximately 40% of novel chemical entities (NCEs) encounter various obstacles during the stages of formulation and development. Drugs categorized as Class II and IV in the Biopharmaceutics Classification System (BCS) frequently exhibit inadequate water solubility, restricted dissolution, and diminished bioavailability. Therefore, the formulation of pharmaceuticals should focus on improving the solubility and dissolution rates of active pharmaceutical ingredients (APIs). As these elements are vital for enhancing bioavailability [2]. Selexipag (SLX) acts as an orally given agonist that targets the prostacyclin (IP) receptor. Upon intake, it is hydrolyzed by carboxylesterase one, yielding an active metabolite that exhibits approximately a 37-fold increase in potency relative to the parent drug. In contrast to other prostanoid receptors (EP1–4, DP, FP, and TP), selexipag and its metabolite exhibit a pronounced selectivity for the IP receptor [3].
Clinically, used for pulmonary arterial hypertension (PAH) treatment, it exerts its effect by significantly raising intracellular cyclic adenosine monophosphate (cAMP) levels, resulting in reduced contraction and proliferation of pulmonary arterial smooth muscle cells [4]. Selexipag minimized likelihood of hospitalization and delays the disease progression risks in PAH patients [5]. Selexipag is a crystalline powder with a pale yellow appearance and is non-hygroscopic. It has pH-dependent solubility and is practically insoluble in water (<1 mg/ml) [6]. It has greater solubility in organic solvents, such as dimethyl sulfoxide (DMSO), methanol, and acetone [7]. The absolute bioavailability of SLX approximately 50%. According to the Biopharmaceutical Classification System (BCS), selexipag belongs to Class II medication, defined by poor solubility and high permeability [8]. However, despite its proven therapeutic efficacy in pulmonary arterial hypertension, the poor aqueous solubility of selexipag remains a major limitation that reduces its oral bioavailability and, consequently, its clinical effectiveness. Therefore, enhancing its solubility is crucial to achieve consistent therapeutic outcomes.
To overcome specific challenges associated with drug formulation, such as poor solubility, we can employ a range of physical and chemical techniques, including prodrug synthesis, salt formation, solid dispersions, hydrotropic substances, solubilizing agents, cosolvent addition, polymorphism investigation, crystal formation, cyclodextrin complexation, lipid formulations, particle size reduction, and nanoformulation methods [9]. Among the different techniques solid dispersion (SD)is considered attention one of the effective formulation strategies to enhance the solubility limitations and subsequently improve the bioavailability of poorly-soluble drugs [10]. Amorphous solid dispersions (ASDs) represent a modern and efficient strategy for enhancing the solubility and oral bioavailability of drugs with limited aqueous solubility [11]. In these systems, the drug is molecularly dispersed within a hydrophilic polymer matrix in the solid state, leading to improved wettability and dissolution performance. These enhancements arise mainly from molecular interactions between the drug and polymer and the amorphous nature of the formulation [12].
The drug is hydrophobic in nature, while the matrix improves dissolution due to its hydrophilic nature [13]. Therefore, the study aims to improve the solubility and dissolution rate of SLX by formulating it as a solid dispersion utilizing multiple methods, specifically kneading and solvent evaporation techniques, with different hydrophilic carriers, including PXM 188, PXM 407, PEG 400, urea, and PEG 600.
MATERIALS AND METHODS
Chemicals and reagents
Selexipag was provided by Hyperchem, China. Polyethylene glycol 6000 (PEG 6000) and Poloxamer 407 (PXM 407) were purchased from Sigma-Aldrich, Germany. Polyethylene glycol 4000 (PEG 4000) was obtained from Loba Chemie, India. Urea was provided by Thomas Baker, India. Poloxamer 188 (PXM 188) was provided by Loba Chemie, India.
Instruments and apparatus
The instruments used in this study included a UV-visible spectrophotometer (Shimadzu, Japan), a dissolution apparatus (Erweka, Germany), and an FTIR spectrophotometer (Shimadzu, Japan). A sonicator (Power Sonica 410, USA), a pH meter (Oakton pH 2100 series, USA), and a hot plate stirrer (Stuart, UK) were also utilized. Additional equipment included a centrifuge (Sigma 3-18K, Germany), an oven (Binder, Germany), a water bath shaker (GFL, Germany), and an electronic balance (Ohaus, Spain).
Design of experiments
The prepared solid dispersion formulations were systematically evaluated using a Design of Experiments (DOE) approach. The DOE included one numerical factor, the carrier-to-drug ratio (1:1,1:3, and 1:5) and two categorical factors: carrier type, including five levels and preparation method, including two levels. A D-optimal response surface design was employed using Design Expert® version 13 to evaluate the influence of selected factors on key responses, including percentage yield, drug content, and saturation solubility. The suggested design was composed of 27 formulations, as illustrated in table 1.
Table 1: Design of experiment for solid dispersion loaded with selexipag
| Formula | A Carrier-to-drug ratio | B Carrier type | C Method |
| 1 | 3 | PXM 188 | Solvent evaporation |
| 2 | 3 | PXM407 | Solvent evaporation |
| 3 | 3 | PEG4000 | Solvent evaporation |
| 4 | 5 | PXM188 | Kneading |
| 5 | 5 | PEG4000 | Kneading |
| 6 | 3 | Urea | Solvent evaporation |
| 7 | 3 | PEG6000 | Kneading |
| 8 | 3 | PXM188 | Kneading |
| 9 | 1 | PXM407 | Kneading |
| 10 | 3 | PEG6000 | Solvent evaporation |
| 11 | 1 | PXM407 | Solvent evaporation |
| 12 | 5 | PEG4000 | Kneading |
| 13 | 1 | PXM188 | Kneading |
| 14 | 1 | PEG4000 | Kneading |
| 15 | 5 | Urea | Solvent evaporation |
| 16 | 3 | PXM407 | Kneading |
| 17 | 5 | PEG6000 | Solvent evaporation |
| 18 | 1 | Urea | Solvent evaporation |
| 19 | 3 | Urea | Kneading |
| 20 | 5 | PXM407 | Solvent evaporation |
| 21 | 1 | PEG6000 | Solvent evaporation |
| 22 | 3 | Urea | Kneading |
| 23 | 3 | PEG4000 | Solvent evaporation |
| 24 | 5 | Urea | Kneading |
| 25 | 3 | PEG6000 | Kneading |
| 26 | 3 | PXM188 | Solvent evaporation |
| 27 | 5 | PXM407 | Kneading |
Note: The numerical values (1, 3, and 5) in the column “Carrier to drug ratio” represent Carrier: Drug ratios of 1:1, 1:3, and 1:5, respectively.
Preparation of selexipag solid dispersion by the solvent evaporation method
In the solvent evaporation method (SE), solid dispersions (SDs) were prepared by accurately weighing selexipag as well as the chosen carrier were each dissolved in ethanol until transparent solutions were achieved. The two solutions were then mixed and magnetically stirred for 45 min to ensure homogeneity. The resultant mixture was poured into Petri dishes and subjected to drying in a hot-air oven at 40 °C for 48 h to ensure the complete elimination of the solvent. The resultant solid material was scraped, crushed, and sieved through a No. 60 screen, then preserved in a moisture-free desiccator until further investigation [14].
Preparation of selexipag solid dispersion by kneading method
In the kneading method (KN), selexipag and various water-soluble carriers, including PXM 407, PXM 188, PEG 6000, PEG 4000, and urea were weighed in various drug-to-polymer ratios (1:1, 1:3, and 1:5) as shown in table 1. The combined materials were moistened with a limited volume of ethanol-water (1:1, v/v) solution and blended completely in a mortar and pestle for 30 min in order to obtain a consistent paste. The produced paste was then dried in a forced air dryer (oven) at 40 °C for 48 h. The dried product was subsequently milled, pulverized, sieved through a No. 60 mesh, after that it was stored in tightly closed containers for further evaluation [15].
Evaluation of selexipag solid dispersion
Determination of percentage yield
The performance of the solvent evaporation and kneading procedures employed in this study was assessed by calculating the actual yield percentage (PY%) of the prepared selexipag solid dispersions. PY% was calculated as the ratio of the measured mass of the resulting solid system to the calculated theoretical mass, according to the following equation [16].
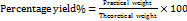 ……. Eq. 1
……. Eq. 1
Determination of drug content
All fabricated SD formulations were subjected to drug content determination. A precisely weighed amount of SLX-SD, equivalent to 5 mg of Selexipag, was dissolved in a 50 ml of ethanol. The mixture was sonicated for 30 min to ensure complete dissolution. The solution was diluted with ethanol following sonication. Subsequently, the UV-visible spectrophotometer operated at its designated λmax (298 nm) to evaluate the drug content of the prepared solution. The equation below was employed to determine the drug content percentage [17].
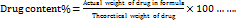 Eq. 2
Eq. 2
Determination of saturation solubility
The saturated solubility of both the prepared selexipag solid dispersion and the pure drug was assessed in phosphate buffer solution (pH 6.8)for each sample, an excess quantity was introduced into plain, tightly capped test tubes, each containing 10 ml of the buffer. For a period of 48 h, the tubes were securely sealed and placed in a water bath shaker that was maintained at 37±0.5 °C to reach equilibrium. After the incubation period (48 h), the samples were centrifuged at 10,000 rpm for 10 min using a Sigma 3-18K centrifuge (Sigma Laborzentrifugen GmbH, Germany) to separate undissolved drug particles. The resulting supernatants were then passed through a 0.45 µm syringe filter. The filtrates were subsequently analyzed by a UV-visible spectrophotometer at a wavelength (305 nm), and all measurements were carried out in triplicate [18].
In vitro dissolution studies
The in vitro dissolution of the produced SLX solid dispersion was assessed and compared to that of pure SLX using a USP type II dissolution apparatus (Erweka DT720 GmbH, Germany). The vessels in the apparatus were filled with phosphate buffer solution (pH 6.8), which served as the dissolution medium in this experiment. An exact quantity of SLX-SD, equivalent to 5 mg of the pure compound, was dispersed in 300 ml of phosphate buffer (pH 6.8) with 1% Brij-35 (to maintain sink condition). The dissolution medium was maintained at 37±0.5 °C under continuous agitation using a paddle operated at 75 rpm.
At predetermined intervals of 10, 20, 30, 45, 60, 90, and 120 min, a sample of 5 ml was withdrawn from the dissolving medium and replaced with an equal volume of fresh phosphate buffer pH 6.8 after each sample collection. Following filtration with a 0.45-micron syringe filter, for each time point, the samples were examined by UV spectrophotometry at λmax (305 nm) to determine the amount of dissolved drug. The concentration of the released drug was then plotted against time to construct the cumulative drug release profile [19].
To assess the difference in the dissolution profiles between the pure drug and the SLX solid dispersion, a statistical comparison was conducted using the similarity factor (f₂), which is calculated using the following equation:
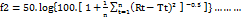 Eq. 3
Eq. 3
The dissolution values of the reference product (Rt) and the test formulation (Tt) are compared across multiple time points (n). In the range of 50 to 100, dissolution patterns are similar when the f2 value is larger than 50. In contrast, profiles with a f2 value below 50 are considered not similar [20].
Fourier transform infrared spectroscopy (FTIR)
Compatibility studies between pure Selexipag, Poloxamer 407 (PXM407), and the selected solid dispersion formulation were carried out using FTIR spectroscopy. Each sample was separately mixed with dry potassium bromide (KBr) and compressed into a transparent disc using a hydraulic press to prepare it for FTIR analysis in transmission mode. The FTIR spectra of the prepared discs were examined using an FTIR spectrophotometer (IRPrestige-21, Shimadzu, Japan) in the range of (400–4000 cm⁻¹) [21].
RESULTS AND DISCUSSION
Analysis of D-optimal design
D-optimal design experiments were conducted to assess the impacts of polymer type, polymer ratio, and preparation process on the responses of selexipag solid dispersions. The design outcomes indicated that all responses were associated with the chosen parameters (table 4). This was indicated by the significant p-values obtained through the analysis of variance (ANOVA) test (tables 2 and 3).
Table 2: ANOVA results for the dependent variables
| Model | R2 | Adjusted R2 | Predicted R2 | Adequate precision | Pvalue | F ratio | ||
| Y1a | Quadratic | 0.9678 | 0.9162 | NA | 15.089 | <0.0001 | 18.77 | Significant |
| Y2b | 2FI | 0.7775 | 0.4740 | NA | 5.869 | 0.0605 | 2.56 | Not Significant |
| Y3c | 2FI | 0.8209 | 0.5767 | NA | 7.8416 | 0.0242 | 3.36 | Significant |
aSaturation solubility, bPercentage yield, cDrug content
Table 3: ANOVA p-values for the effects of ratio, carrier type, and method on yield, drug content, and solubility
| Response | A(polymer: drug ratio) | B (carrier type) | C (method) |
| Yield% | 0.2862 | 0.3214 | 0.0083 |
| Drug content% | 0.0066 | 0.0369 | 0.0369 |
| Saturation solubility | <0.0001 | <0.0001 | <0.0001 |
Percentage of yield and drug content
The optimal technique for preparing solid dispersion was examined by calculating the percentage yield (PY%) of the produced SLX-solid dispersion. As shown in Table4, both the solvent evaporation and kneading methods produced acceptable PY percentages ranging from 93.16% to 97.27%. The results demonstrated good comparability of the solvent evaporation and kneading methods. Based on these results, both methods were considered appropriate and effective. Furthermore, in all prepared formulations, the amount of SLX loading in the solid dispersions ranged from 95.7% to 102.5%, suggesting a uniform distribution of SLX particles in all the prepared formulation.
Saturation solubility
All prepared selexipag solid dispersion formulations exhibited a significant enhancement in saturation solubility (p<0.05) when assessesed in comparison with the pure drug, as shown in table 4. In phosphate buffer (pH 6.8), the saturation solubility of selexipag was approximately 1.5 µg/ml±0.04, indicating that the pure drug is practically insoluble in aqueous medium [6]. The improvement in solubility can be influenced by several factors, including the preparation method, the type of polymer used, and the drug-to-polymer ratio [22]. In general, the enhancement in solubility may be attributed to improved drug wettability, reduction in particle size, the hydrophilic features of the carriers and the enhanced porosity of the solid dispersions [23, 24].
In terms of method effects, the solvent evaporation approach produced the most soluble solid dispersion. This observation agrees with recent findings by Ali et al. (2024), who demonstrated that the solvent evaporation method yielded superior solubility enhancement compared to kneading, owing to improved drug-polymer miscibility and uniform molecular dispersion [25]. This could be as a result of the solvent evaporation method producing a more homogeneous molecular dispersions of the drug in the hydrophilic carrier matrix compared with the kneading method [26]. Additionally, solubility is significantly impacted by the type of carrier. PXM407 has the greatest improvement in solubility among the carriers that are being used. This may be attributed to the increased solubilizing capacity and the high hydrophilic–lipophilic balance (HLB = 22 at 22 °C) of the copolymer surfactant, which makes it highly water-soluble carrier [27]. Similar improvement in solubility was reported by Pironi et al. (2023), where Poloxamer 407-based solid dispersions showed enhanced drug solubility and stability compared to physical mixtures [28].
Moreover, the solubility of SLX was affected by the drug-to-polymer ratio, when an increased in the polymer ratio resulted in higher solubility. This finding agrees with Aldeeb et al. (2023), who observed that increasing the polymer concentration significantly improved the solubility of mirtazapine solid dispersions due to the hydrophilic nature of the polymers [29]. However, an exception was observed in two formulations (F20 and F27) that contain PXM407:SLX at a 5:1 ratio, prepared by solvent evaporation and kneading methods, respectively, where increasing the polymer ratio resulted in a decrease in solubility. This may be due to increased viscosity of polymer matrix, which could hinder the diffusion of drug into the aqueous medium [30]. The solubility results of the 27 prepared formulations are summarized in table 4.
The polynomial equation in terms of coded factors for the inverse of saturation solubility was as follows:
1/(Saturation Solubility) =+0.2746 − 0.1182A+0.1339B₁ − 0.1759B₂+0.0148B₃ − 0.0132B₄+0.0775C − 0.0674AB₁+0.0118AB₂ − 0.0339AB₃+0.0537AB₄ − 0.0422AC+0.0341B₁C − 0.0417B₂C − 0.0249B₃C+0.0333B₄C+0.0519A²
where:
A = polymer-to-drug ratio,
B = polymer type,
C = preparation method.
Positive coefficients indicate an increase in solubility, while negative coefficients show a decrease. The negative value of factor A (polymer-to-drug ratio) means that raising the ratio tends to lower the solubility, which was particularly evident in formulations containing PXM407. In contrast, other polymers did not show a noticeable reduction in solubility with increasing ratio. The positive signs of factors B (polymer type) and C (method) demonstrate that both the type of carrier and the preparation method contributed to higher solubility values. This confirms that both the choice of carrier and the preparation technique play a major role in improving the wettability and molecular dispersion of selexipag within the solid dispersion matrix.
Table 4: Effect of factors, carrier to drug ratio(A), carrier type(B), and method (C)On responses: yield percentage, drug content percentage and saturation solubility
| Formula | Carrier to drug ratio | Carrier type | Method | Percentage yield % | % of drug content |
Saturation solubility (µg/ml) |
| 1 | 3 | PXM188 | Solvent evaporation | 94.12% | 98.8%±0.02 | 7.46±0.01 |
| 2 | 3 | PXM407 | Solvent evaporation | 95.4% | 99.45±0.01 | 25.9±0.2 |
| 3 | 3 | PEG4000 | Solvent evaporation | 93.4% | 98.83%±0.02 | 3.79±0.21 |
| 4 | 5 | PXM188 | Kneading | 96.5% | 99.5%±0.07 | 3.34±0.16 |
| 5 | 5 | PEG4000 | Kneading | 97.27% | 102.5%±0.009 | 3.11±0.16 |
| 6 | 3 | Urea | Solvent evaporation | 93.3% | 100.4%±0.02 | 3.37±0.57 |
| 7 | 3 | PEG6000 | Kneading | 96% | 99%±0.06 | 2.99±0.4 |
| 8 | 3 | PXM188 | Kneading | 96.9% | 99.1%±0.04 | 2.45±0.43 |
| 9 | 1 | PXM407 | Kneading | 95.5% | 99.85%±0.02 | 2.69±0.14 |
| 10 | 3 | PEG6000 | Solvent evaporation | 93.7% | 100 %±0.06 | 3.35±0.58 |
| 11 | 1 | PXM407 | Solvent evaporation | 96.4% | 95.7%±0.02 | 5.18±0.67 |
| 12 | 5 | PEG4000 | Kneading | 97% | 101.6%±0.01 | 3.16±0.38 |
| 13 | 1 | PXM188 | Kneading | 96% | 97.58%±0.004 | 1.95±0.008 |
| 14 | 1 | PEG4000 | Kneading | 93.16 % | 99.1±0.08 | 1.76±0.077 |
| 15 | 5 | Urea | Solvent evaporation | 94.2% | 98.99%±0.01 | 4.82±0.33 |
| 16 | 3 | PXM407 | Kneading | 96.4% | 100.3%±0.04 | 17.53±0.91 |
| 17 | 5 | PEG600 | Solvent evaporation | 94.4% | 100.4%±0.1 | 6.76±0.98 |
| 18 | 1 | Urea | Solvent evaporation | 95.6% | 98.7%±0.07 | 2.04±0.2 |
| 19 | 3 | Urea | Kneading | 96.5% | 96.4%±0.04 | 1.93±0.057 |
| 20 | 5 | PXM407 | Solvent evaporation | 95.7% | 101.4%±0.03 | 16.49±0.52 |
| 21 | 1 | PEG6000 | Solvent evaporation | 94.3% | 96.28%±0.08 | 2.72±0.03 |
| 22 | 3 | Urea | Kneading | 96.6% | 96.2%±0.08 | 1.90±0.1 |
| 23 | 3 | PEG4000 | Solvent evaporation | 96.3% | 100.5%±0.1 | 4.70±0.91 |
| 24 | 5 | Urea | Kneading | 96% | 96.6%±0.06 | 2.94±0.85 |
| 25 | 3 | PEG6000 | Kneading | 94.8% | 98.73%±0.05 | 2.86±0.76 |
| 26 | 3 | PXM188 | Solvent evaporation | 95% | 98.78%±0.06 | 5.99±0.64 |
| 27 | 5 | PXM407 | Kneading | 95.7% | 97.4%±0.01 | 12.7±0.96 |
*Abbreviations: PXM = Poloxamer, PEG = Polyethylene glycol. value are expressed as mean±standard deviation (n = 3).
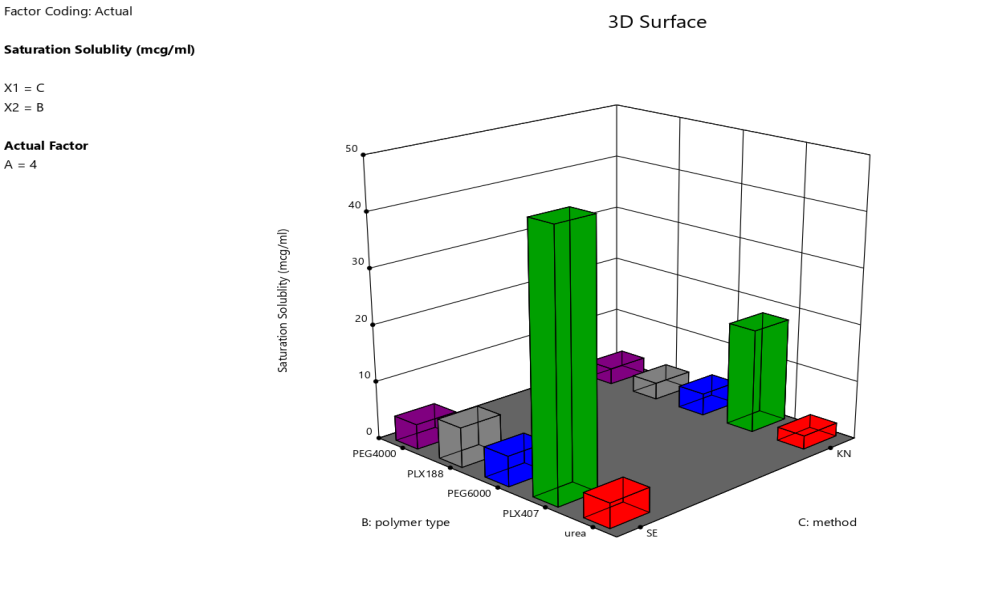
Fig. 1: 3D surface plot showing the interactive effects of polymer type (B) and preparation method (C) on the saturation solubility of selexipag solid dispersions
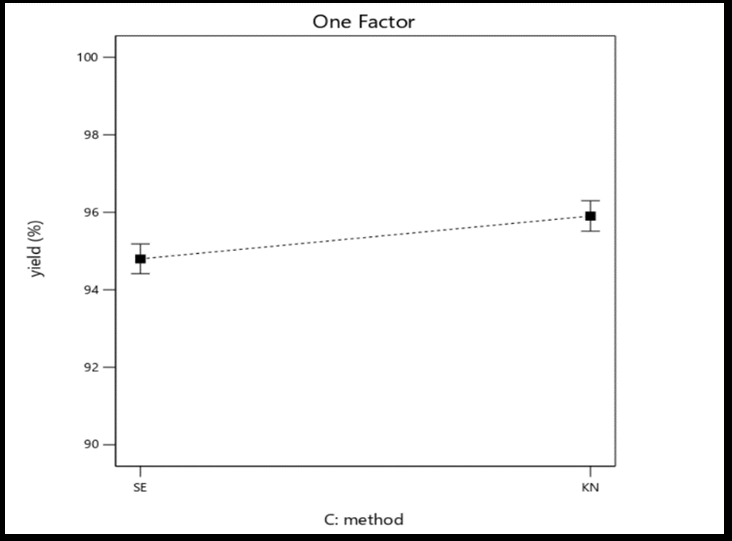
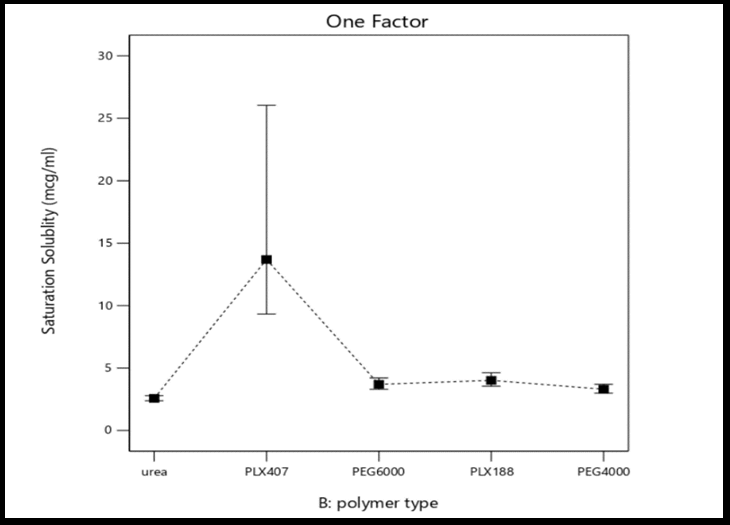
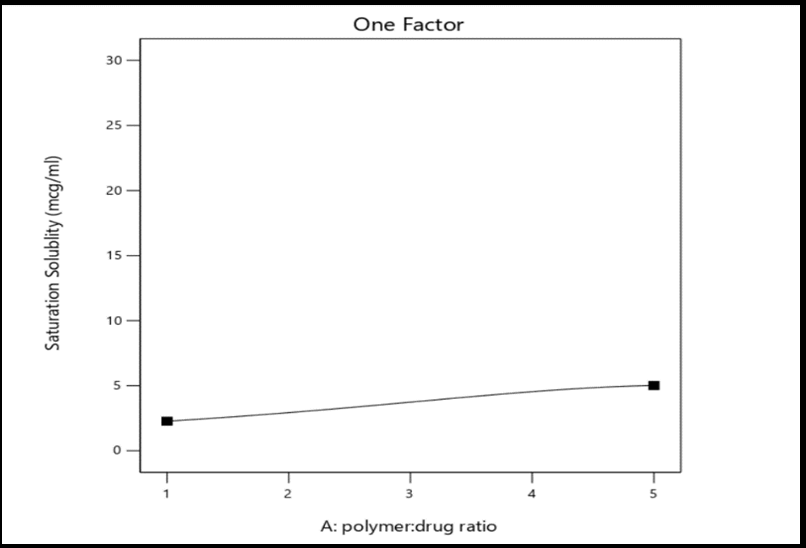
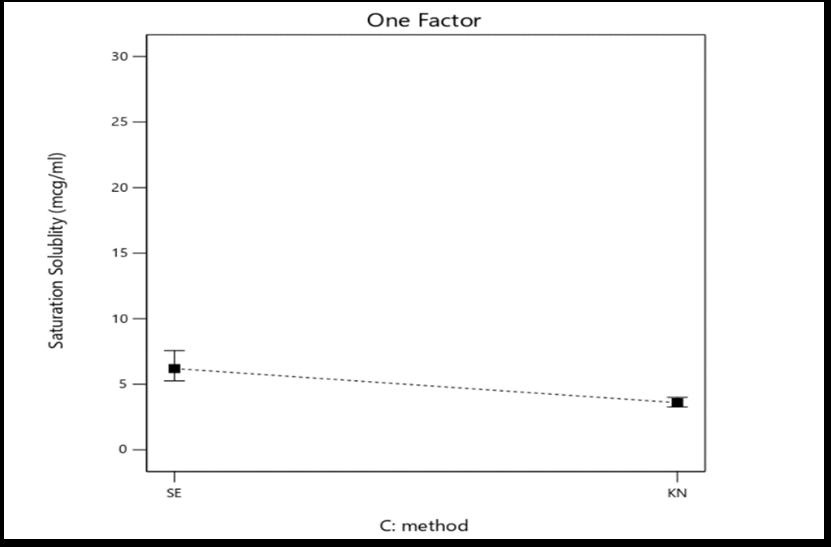
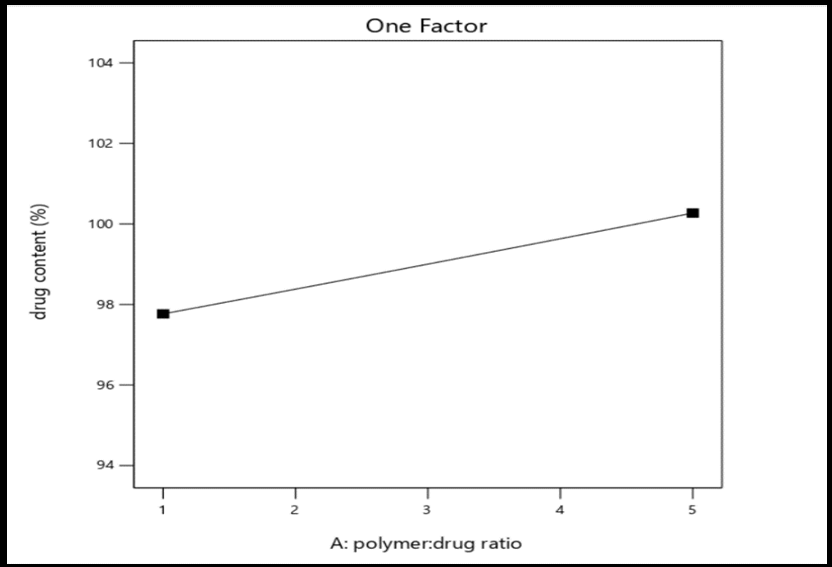
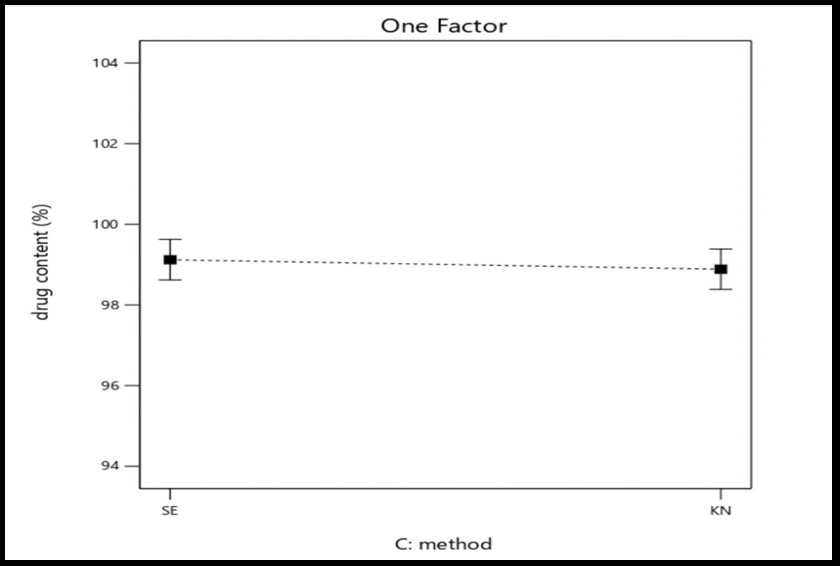
Fig. 2: Graphic model illustrating the one-factor effects of polymer-to-drug ratio (A), polymer type (B), and preparation method (C) on the percentage yield, drug content, and saturation solubility of selexipag solid dispersions
In vitro dissolution studies
According to the results obtained from the apparent solubility study, the formulations that demonstrated relatively improved solubility (F1, F2, F3, F6, F10, F16, and F20) were selected to compare their in vitro dissolution behavior. These formulations were analyzed to investigate the impact of polymer type, drug-to-polymer ratio, and solid dispersion preparation method on the in vitro dissolution profile of selexipag.
Effect of polymer type
Formulations containing different polymer types, including PXM188 (F1), PXM407 (F2), PEG4000 (F3), urea (F6), and PEG6000 (F10), were selected to investigate the effect of polymer type at a fixed drug-to-polymer proportion (1:3), prepared via the solvent evaporation technique. Fig. 3 demonstrated that the dissolution rate was enhanced by all of the previously mentioned formulations compared with the pure SLX drug, with the following f₂ values (relative to the pure drug): 35.62, 17.23, 37.16, 46.31, and 39.82, respectively.
After 30 min, PXM407 (F2) achieved the highest drug release profile (88%). In contrast, the other formulations (F1, F3, F6, F10) and the pure drug exhibited lower release values of 54%, 56%, 49.3%, 57%, and 38%, respectively, showing a statistically significant difference (p<0.05) relative to the pure drug. The enhancement in dissolution profile by PXM407 can be attributed to the amphiphilic structure of PXM 407, which consists of a sequence of ethylene oxide (EO) blocks in conjunction with propylene oxide (PO) blocks. These characteristic blocks enable PXM 407 to self-assemble into micelles when contact with water. These monomolecular micelles can assemble into aggregates of varied sizes, thereby enhancing drug solubilization [31].
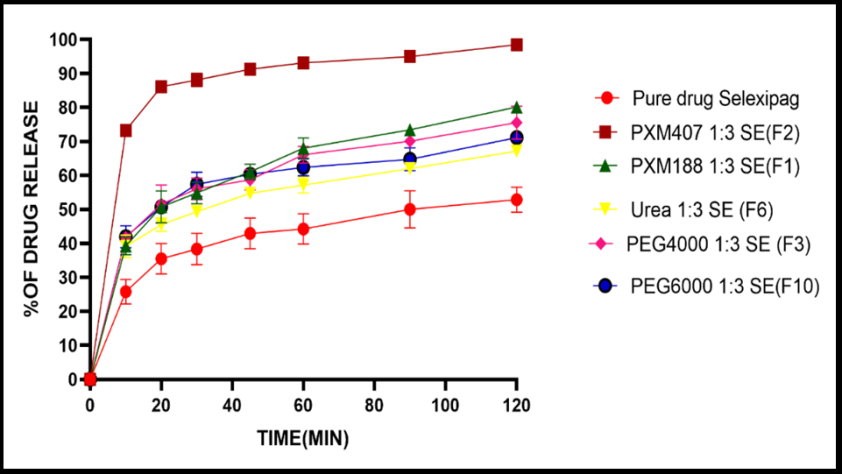
Fig. 3: Effect of carrier type on the dissolution profile of selexipag solid dispersions prepared by solvent evaporation (SE) method at a fixed 1:3 drug-to-polymer ratio. Values are presented as the mean±standard deviation, in dissolution medium composed of phosphate buffer (pH 6.8) at 37 °C with experiments performed in triplicate (n=3). PXM = Poloxamer, PEG = Polyethylene glycol, SE = solvent evaporation
Effect of polymer ratio
Solid dispersions of PXM407 produced using an identical preparation method (solvent evaporation) with different drug: polymer proportions (1:3 and 1:5) (F2, F20), respectively, were considered for evaluating the influence of drug: polymer ratio on the in vitro dissolution behavior of selexipag. Formula 2 demonstrated a substantial enhancement in the dissolution rate at a ratio of 1:3 (f₂ = 17.23), releasing 88% of the drug after 30 min. However, further increasing the polymer concentration to a 1:5 drug-to-polymer ratio (F20) resulted in retardation of drug release (59% after 30 min, f₂ = 31.34), as shown in fig. 4. This may be attributed to the formation of a viscous matrix that limited drug diffusion [32].
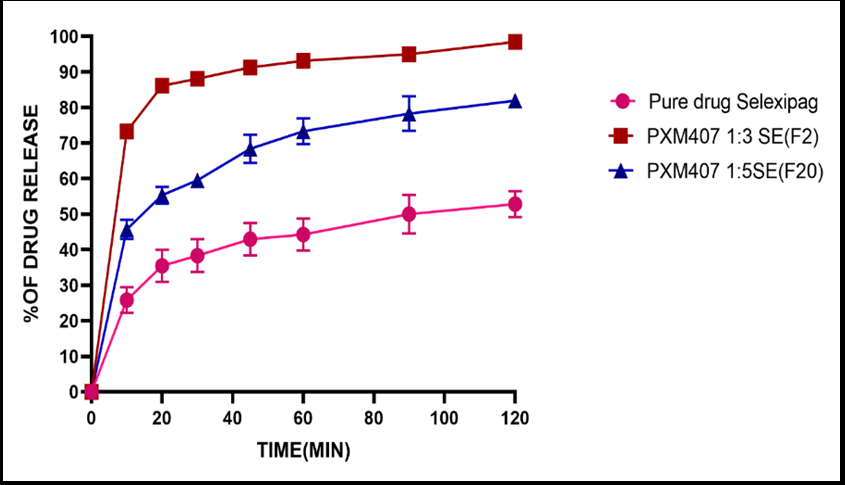
Fig. 4: Effect of the drug-to-polymer ratio on the dissolution profile of selexipag solid dispersions prepared by the solvent evaporation method. Each point represents the mean±SD of three determinations (n = 3). Experiments were conducted at 37 °C in phosphate buffer (pH 6.8)
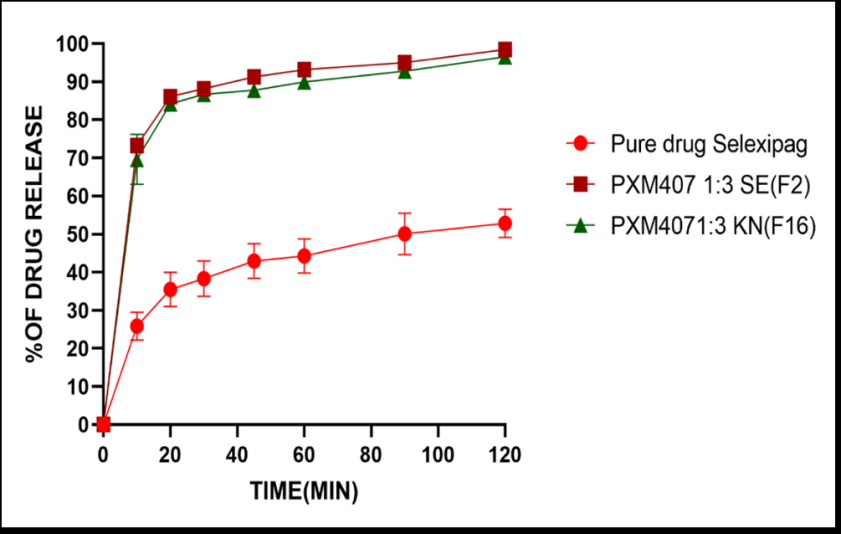
Fig. 5: Impact of the preparation method on the in vitro dissolution profile of selexipag solid dispersions formulated with PXM407 at a 1:3 drug-to-carrier ratio. Data are expressed as mean±SD (n = 3). Experiments were carried out in phosphate buffer (pH 6.8) at 37 °C
Effect of preparation method
The dissolution behavior of PXM407 SDs prepared at a 1:3 drug-to-carrier ratio using two different methods SE and KN represented by F2 and F16, showed comparable release profiles, with no significant differences observed at all time points between the two methods (f₂ = 17.23 for F2 and 18.34 for F16, relative to the pure SLX drug). Both formulations released 88% and 86.7% of the drug after 30 min, respectively, and the release profile remained consistent throughout the study, as illustrated in fig. 5.
Table 5: Best-fit kinetic models with mechanism interpretation
| Formulation | Bes-fit model | R² value | k (K-P) | Mechanism interpretation |
| Pure SLX | Korsmeyer–Peppas | 0.994 | 0.144 | Fickian diffusion |
| PXM407 SE 1:5 | Korsmeyer–Peppas | 0.9994 | 24.249 | Fickian diffusion |
| PEG6000 SE 1:3 | Korsmeyer–Peppas | 0.9962 | 25.102 | Fickian diffusion |
| PEG4000 SE 1:3 | Korsmeyer–Peppas | 0.9967 | 22.764 | Fickian diffusion |
| Urea SE 1:3 | Korsmeyer–Peppas | 0.9997 | 23.806 | Fickian diffusion |
| PXM407 SE 1:3 | Korsmeyer–Peppas | 0.9966 | 57.172 | Fickian diffusion |
| PXM407 KN 1:3 | Korsmeyer–Peppas | 0.9937 | 55.139 | Fickian diffusion |
| PXM188 SE 1:3 | Korsmeyer–Peppas | 0.9983 | 20.202 | Fickian diffusion |
All selexipag solid dispersion formulations and the pure drug exhibited the best fit with the Korsmeyer–Peppas model (R² = 0.9937–0.9997), as this model showed the lowest Residual Sum of Squares and Reduced Chi-square values among all evaluated kinetic models. The diffusion exponent values (n<0.45) confirmed a Fickian diffusion mechanism [33], indicating that drug release was governed primarily by diffusion through the hydrated polymeric matrix. The similarity of the release mechanism among all formulations, including those containing PEGs, Poloxamers, and urea, indicates that selexipag release was mainly governed by diffusion through the hydrated polymeric matrices. The higher release rate constants (k) of the solid dispersions compared with the pure drug demonstrate the enhanced dissolution performance achieved through improved wettability and reduced crystallinity induced by the hydrophilic carriers.
Selection of the optimum formula
Formula 2, which contain PXM407: selexipag in 3:1 ratio was selected as the optimum formula and subjected to FTIR investigation. This is because it had the highest solubility and the highest amount of released drug after 30 min compared with the other formulations.
Fourier transforms infrared spectroscopy (FTIR)
Fourier Transform Infrared (FTIR) spectroscopy was employed to evaluate the potential molecular interactions between selexipag and Poloxamer 407, and to confirm the successful incorporation of the drug within the polymer matrix.
The FTIR spectrua of pure drug selexipag, PXM407 and the selected formula are shown in fig. (6-8). The pure selexipag exhibited several characteristic absorption bands consistent with its functional groups, as summarized in table 6.
These characteristic peaks are in agreement with previously reported spectra of selexipag [6]. While FTIR spectrum of PXM407 display the characteristic peaks at 3446.79 cm,⁻¹2966.52-2883.58 cm⁻¹,1111.00 cm⁻¹, corresponding to O-H streching, C-H aliphatic streteching, C-O-C streching respectively. This obsorvation is consistent with previous reports [34].
Table 6: Characteristic FTIR absorption peaks of pure SLX
| Functional group assignment | Wavenumber (cm-1) |
| C=O stretching | 1726.29 |
| Aromatic C=C stretching | 1562.34-1481.33 |
| N-H stretching | 3419.79 |
| Aromatic C-H stretching | 3026.31 |
| Aliphatic C-H stretching | 2937.59 – 2870.08 |
| C-O-C stretching | 1124.50 |
| C=N stretching | 1672.28 |
SO₂ stretching C-N stretching |
1330.88 1242.16 |
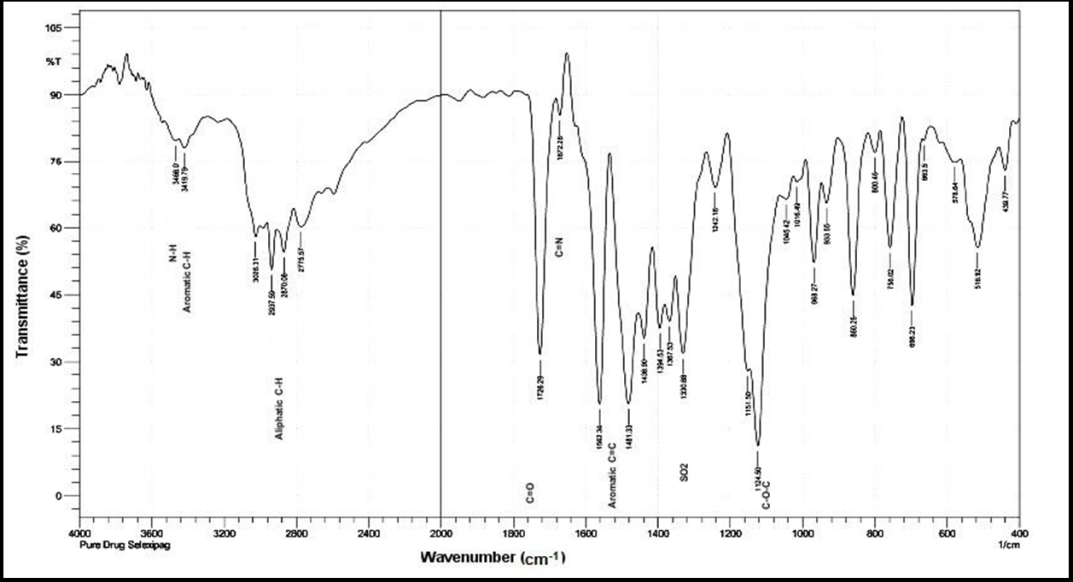
Fig. 6: FTIR spectrum of pure drug (selexipag)
In comparison, the FTIR spectrum of the selexipag-loaded Poloxamer 407 formulation revealed several significant spectral changes. The carbonyl stretching peak (1722 cm⁻¹) exhibited a noticeable shift and reduction in intensity, suggesting the involvement of this group in hydrogen bonding with functional groups within the polymer. Furthermore, the broadening and intensification of the O–H stretching band near 3444.87 cm⁻¹ indicate the formation of enhanced hydrogen bonding networks between selexipag and the hydrophilic polyethylene oxide (PEO) segments of Poloxamer 407 [35]. Several distinctive peaks present in the spectrum of pure selexipag were either diminished or overlapped in the formulation, supporting the hypothesis that the drug was not simply physically mixed, but rather molecularly dispersed within the polymer matrix. However, no additional peak was observed in any binary system, indicating absence of any chemical interaction between selexipag and polymer.
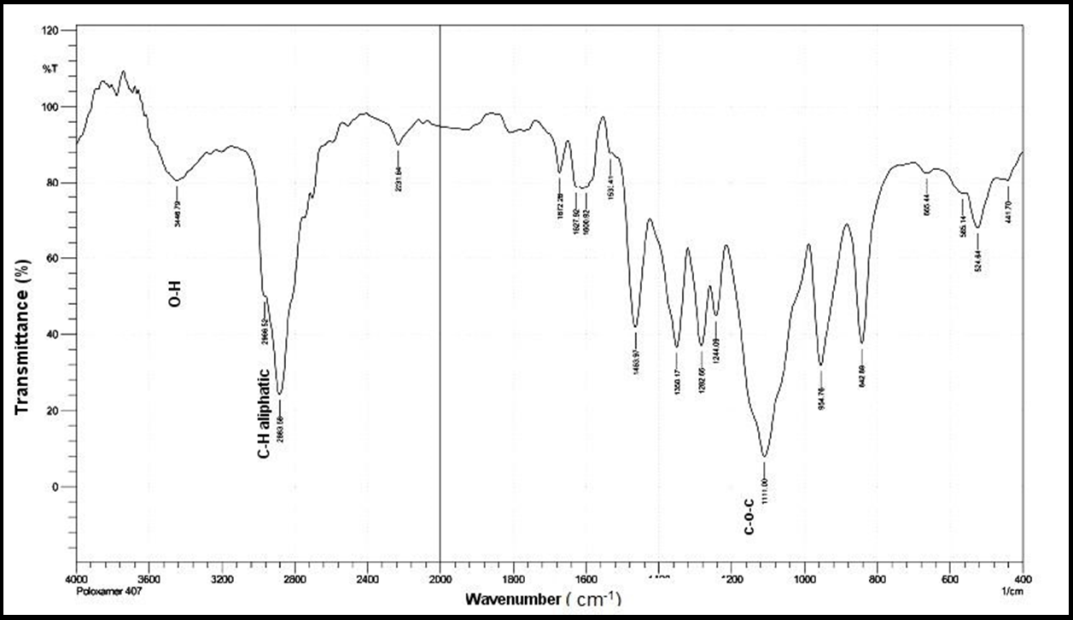
Fig. 7: FTIR spectrum of poloxamer 407
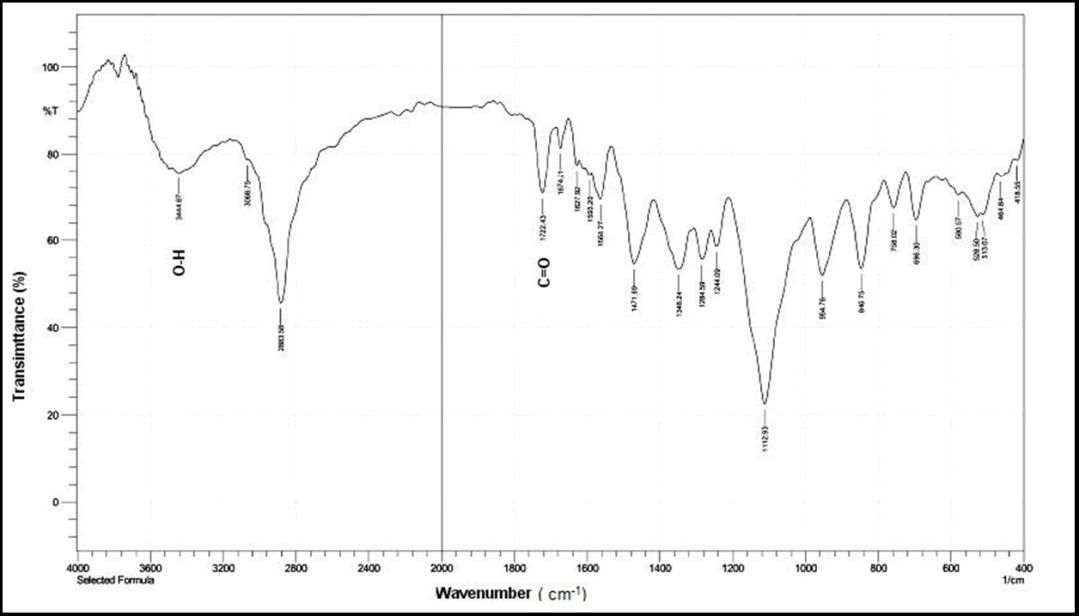
Fig. 8: FTIR spectrum of selected formula (F2)
CONCLUSION
Based on the findings of this research, it can be concluded that the solubility of selexipag was significantly improved through the application of the solid dispertion approach. The Design of Experiment (DoE) analysis revealed that the carrier type was the most significant factor influencing solubility, followed by the drug-to-carrier ratio and the preparation method. The solubility and dissolution rate of the final product were significantly influenced by these formulation variables and processing parameters. Among the tested carriers, Poloxamer 407 exhibited the most pronounced enhancement, with the formulation prepared by the solvent evaporation method at a 1:3 drug-to-polymer ratio achieving the highest solubility and an 88% dissolution rate within 30 min. Overall, solid dispersion represents an effective method to enhance both the solubility and dissolution rate of poorly water-soluble drugs. Future studies should focus on evaluating the stability of the amorphous form, performing further solid-state characterization (DSC and PXRD), and investigating the in vivo pharmacokinetic behavior of the optimized formulation.
ACKNOWLEDGEMENT
The authors would like to express their gratitude to the department of pharmaceutics and the faculty of pharmacy at the University of Kufa for providing the necessary facilities for the successful completion of this work.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
All authors contributed equally to the development and execution of this research. Each author was actively involved in the conceptualization, data acquisition, analysis, and manuscript. The final manuscript has been thoroughly reviewed and approved by all authors prior to publication.
CONFLICT OF INTERESTS
Declared none
REFERENCES
Liu X, Zhao L, Wu B, Chen F. Improving solubility of poorly water-soluble drugs by protein-based strategy: a review. Int J Pharm. 2023;634:122704. doi: 10.1016/j.ijpharm.2023.122704, PMID 36758883.
Bhalani DV, Nutan B, Kumar A, Singh Chandel AK. Bioavailability enhancement techniques for poorly aqueous soluble drugs and therapeutics. Biomedicines. 2022;10(9):2055. doi: 10.3390/biomedicines10092055, PMID 36140156.
Dubey R, Pothuvan U, Bhamare P, Singh A, Upmanyu N. A review on drug of pediatric pulmonary arterial hypertension (PAH), their chemistry and pharmaceutical dosage forms. J Drug Delivery Ther. 2020;10(2-s):156-70. doi: 10.22270/jddt.v10i2-s.3947.
Duggan ST, Keam SJ, Burness CB. Selexipag: a review in pulmonary arterial hypertension. Am J Cardiovasc Drugs. 2017;17(1):73-80. doi: 10.1007/s40256-016-0209-9, PMID 27988834.
Noel ZR, Kido K, Macaulay TE. Selexipag for the treatment of pulmonary arterial hypertension. Am J Health Syst Pharm. 2017;74(15):1135-41. doi: 10.2146/ajhp160798, PMID 28533253.
Alwan RM, Rajab NA. Nanosuspensions of selexipag: formulation characterization and in vitro evaluation. Iraqi J Pharm Sci. 2021;30(1):144-53. doi: 10.31351/vol30iss1pp144-153.
Chandramore KR, Sonawane SS, Ahire RS, Nagidi HR, Ahire SB, Jadhav PB. Development and validation of a stability-indicating LC method for selexipag: in silico toxicity study and characterization of its degradation products. Chem Methodol. 2025;9(2):427-47. doi: 10.48309/chemm.2025.507044.1903.
Attia MS, Ghazy FE. Ameliorating the poor dissolution rate of selexipag in aqueous acidic conditions following confinement into mesoporous silica. Ind J Pharm Edu Res. 2023;57(4):1002-11. doi: 10.5530/ijper.57.4.122.
Nyamba I, Sombie CB, Yabre M, Zime Diawara H, Yameogo J, Ouedraogo S. Pharmaceutical approaches for enhancing solubility and oral bioavailability of poorly soluble drugs. Eur J Pharm Biopharm. 2024;204:114513. doi: 10.1016/j.ejpb.2024.114513, PMID 39313163.
Chang CW, Wong CY, Wu YT, Hsu MC. Development of a solid dispersion system for improving the oral bioavailability of resveratrol in rats. Eur J Drug Metab Pharmacokinet. 2017;42(2):239-49. doi: 10.1007/s13318-016-0339-0, PMID 27118361.
Bhujbal SV, Mitra B, Jain U, Gong Y, Agrawal A, Karki S. Pharmaceutical amorphous solid dispersion: a review of manufacturing strategies. Acta Pharm Sin B. 2021;11(8):2505-36. doi: 10.1016/j.apsb.2021.05.014, PMID 34522596.
Sharma B. Drugs dissolution enhancement by using solid dispersion technique: a review. EPRA Int J of Multidisciplinary Research. 2021 Jan;7(1):1-2. doi: 10.36713/epra2013.
Malkawi R, Malkawi WI, Al-Mahmoud Y, Tawalbeh J. Current trends on solid dispersions: past present and future. Adv Pharmacol Pharm Sci. 2022;2022:5916013. doi: 10.1155/2022/5916013, PMID 36317015.
Mohana M, Vijayalakshmi S. Development and characterization of solid dispersion-based orodispersible tablets of cilnidipine. Beni-Suef Univ J Basic Appl Sci. 2022 Dec 1;11(1):83-94. doi: 10.1186/s43088-022-00259-3.
Borawake PD, Arumugam K, Shinde JV. Formulation of solid dispersions for enhancement of solubility and dissolution rate of simvastatin. Int J Pharm Pharm Sci. 2021;13(7):94-100. doi: 10.22159/ijpps.2021v13i7.41205.
Alezzy AA, Al-Khedairy EB. Preparation and evaluation of aceclofenac solid dispersion by fusion technique and effervescent-assisted fusion technique: comparative study. Res J Pharm Technol. 2023;16(11):5358-65. doi: 10.52711/0974-360X.2023.00868.
Sehgal N, Gupta NV, DV G, PS. Fabrication and evaluation of solid dispersion containing glibenclamide. Asian J Pharm Clin Res. 2018;11(8):158. doi: 10.22159/ajpcr.2018.v11i8.26236.
Elsegaie D, El-Nabarawi MA, Mahmoud HA, Teaima M, Louis D. A comparative study on cyclodextrin derivatives in improving oral bioavailability of etoricoxib as a model drug: formulation and evaluation of solid dispersion-based fast-dissolving tablets. Biomedicines. 2023;11(9):2440. doi: 10.3390/biomedicines11092440, PMID 37760881.
Abdul-Hasan MT, Al-Shaibani AJ, Wannas AN, Hasan Al-gburi KM. Quality control testing of conventional clopidogrel bisulfate tablets marketed in Iraq. Int J App Pharm. 2022;14(1):221-5. doi: 10.22159/ijap.2022v14i1.43331.
Muselik J, Komersova A, Kubova K, Matzick K, Skalicka B. A critical overview of FDA and EMA statistical methods to compare in vitro drug dissolution profiles of pharmaceutical products. Pharmaceutics. 2021 Oct 15;13(10):1703. doi: 10.3390/pharmaceutics13101703, PMID 34683995.
Md S, Mehboob SZ, Doddayya H. Preparation and characterization of fluconazole topical nanosponge hydrogel. Int J Pharm Pharm Sci. 2024;16(4):18-26. doi: 10.22159/ijpps.2024v16i4.50589.
Ali MH, Ahmed KK. Solubility and dissolution rate enhancement of bilastine by solid dispersion technique. Iraqi J Pharm Sci. 2025;34(1):218-29. doi: 10.31351/vol34iss1pp218-229.
Tekade AR, Yadav JN. A review on solid dispersion and carriers used therein for solubility enhancement of poorly water-soluble drugs. Adv Pharm Bull. 2020;10(3):359-69. doi: 10.34172/apb.2020.044, PMID 32665894.
Nair AR, Lakshman YD, Anand VS, Sree KS, Bhat K, Dengale SJ. Overview of extensively employed polymeric carriers in solid dispersion technology. AAPS PharmSciTech. 2020;21(8):309. doi: 10.1208/s12249-020-01849-z, PMID 33161493.
Ali IS, Sajad UA, Abdul Rasool BK. Solid dispersion systems for enhanced dissolution of poorly water-soluble candesartan cilexetil: in vitro evaluation and simulated pharmacokinetics studies. PLOS One. 2024;19(6):e0303900. doi: 10.1371/journal.pone.0303900, PMID 38843120.
Elmubarak EH, Osman ZA, Abdelrahman M. Formulation and evaluation of solid dispersion tablets of furosemide using polyvinylpyrrolidone K-30. Int J Curr Pharm Sci. 2021;13(2):43-50. doi: 10.22159/ijcpr.2021v13i2.41554.
Szafraniec J, Antosik A, Knapik Kowalczuk J, Chmiel K, Kurek M, Gawlak K. Enhanced dissolution of solid dispersions containing bicalutamide subjected to mechanical stress. Int J Pharm. 2018 May 5;542(1-2):18-26. doi: 10.1016/j.ijpharm.2018.02.040, PMID 29481948.
Pironi AM, Eloy JO, Rodero CF, Antonio SG, Alonso JD, Chorilli M. PVP solid dispersions containing poloxamer 407 or TPGS for the improvement of ursolic acid release. Braz J Pharm Sci. 2023;59:e21217. doi: 10.1590/s2175-97902023e21217.
Aldeeb RA, Mahdy MA, El-Nahas HM, Musallam AA. Design of mirtazapine solid dispersion with different carriers systems: optimization in vitro evaluation and bioavailability assessment. Drug Deliv Transl Res. 2023;13(9):2340-52. doi: 10.1007/s13346-023-01316-9, PMID 36940079.
Ricci EJ, Lunardi LO, Nanclares DM, Marchetti JM. Sustained release of lidocaine from poloxamer 407 gels. Int J Pharm. 2005;288(2):235-44. doi: 10.1016/j.ijpharm.2004.09.028, PMID 15620863.
Fakhruldeen ZH, Abdul Hasan MT. Formulation and characterization of isradipine solid dispersion with enhanced solubility. Int J App Pharm. 2025;17(1):439-45. doi: 10.22159/ijap.2025v17i1.52230.
Kolasinac N, Kachrimanis K, Homsek I, Grujic B, Duric Z, Ibric S. Solubility enhancement of desloratadine by solid dispersion in poloxamers. Int J Pharm. 2012;436(1-2):161-70. doi: 10.1016/j.ijpharm.2012.06.060, PMID 22772487.
Zhu W, Long J, Shi M. Release kinetics model fitting of drugs with different structures from viscose fabric. Materials (Basel). 2023 Apr 21;16(8):3282. doi: 10.3390/ma16083282, PMID 37110118.
Eloy JO, Marchetti JM. Solid dispersions containing ursolic acid in poloxamer 407 and PEG 6000: a comparative study of fusion and solvent methods. Powder Technol. 2014;253(1):98-106. doi: 10.1016/j.powtec.2013.11.017.
Eloy JO, Saraiva J, Albuquerque S, Marchetti JM. Preparation characterization and evaluation of the in vivo trypanocidal activity of ursolic acid-loaded solid dispersion with poloxamer 407 and sodium caprate. Braz J Pharm Sci. 2015;51(1):101-9. doi: 10.1590/S1984-82502015000100011.