
Int J App Pharm, Vol 18, Issue 2, 2026, 173-179Original Article
COMPUTATIONAL INSIGHTS INTO THE ALLOSTERIC INHIBITION OF BACE1 BY SULFORAPHANE: A MOLECULAR DOCKING AND DYNAMICS STUDY
MITSUE OKA1,4, MALA HIKMAWAN PRIMANA1,4, BONIFACIUS IVAN WIRANATA2, ENADE PERDANA ISTYASTONO3, FLORENTINUS DIKA OCTA RISWANTO4*
1Magister of Pharmacy Study Program, Faculty of Pharmacy, Sanata Dharma University, Campus 3 Paingan, Maguwoharjo, Depok, Sleman, Yogyakarta 55282 Indonesia. 2Pharmaceutical Sciences Department, Faculty of Pharmacy, Widya Mandala Catholic University, Surabaya, Indonesia. 3Research Center for Cheminformatics and Molecular Modeling, Department of Pharmacy, School of Medicine and Health Sciences, Atma Jaya Catholic University of Indonesia, Jl. Pluit Raya No. 2, Jakarta Utara, DKI Jakarta, 14440, Indonesia. 4Research Group of Computer-Aided Drug Design and Discovery of Bioactive Natural Products, Sanata Dharma University, Yogyakarta 55282, Indonesia
*Corresponding author: Florentinus Dika Octa Riswanto; *Email: dikaocta@usd.ac.id
Received: 10 Nov 2025, Revised and Accepted: 05 Feb 2026
ABSTRACT
Objective: This study aimed to investigate the allosteric binding site and dynamic stability of the sulforaphane-BACE1 complex to provide structural insights into its selective inhibition mechanism.
Methods: Molecular docking was performed using YASARA Structure, followed by five independent 10 ns molecular dynamics simulations conducted with GROMACS to evaluate the stability of the sulforaphane–BACE1 complex. Key interactions and stability parameters were analyzed using RMSD-based metrics and interaction fingerprint profiling.
Results: Global docking identified 35 possible binding pockets, with one predominant allosteric site showing the most favorable binding energy and clustering. Molecular dynamics simulations revealed that sulforaphane maintained stable orientations in two of five trajectories (R2 and R4), indicating replica-dependent stability consistent with moderate affinity and allosteric flexibility. Interaction analysis highlighted persistent hydrophobic contacts with ALA173, LEU172, PRO313, GLU315, and ASP323 as major stabilizing residues.
Conclusion: Sulforaphane demonstrates moderate but reproducible allosteric binding to BACE1, primarily stabilized by hydrophobic interactions and key polar contacts. These findings establish a structural model supporting sulforaphane’s selective modulation of BACE1 and provide a computational framework for designing improved allosteric inhibitors for Alzheimer’s disease therapy.
Keywords: Alzheimer’s disease, BACE1, Sulforaphane, Molecular dynamics, Allosteric inhibitor
© 2026 The Authors.Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijap.2026v18i2.57436 Journal homepage: https://innovareacademics.in/journals/index.php/ijap
INTRODUCTION
Alzheimer’s disease is a progressive neurodegenerative disorder and the leading cause of dementia in the global elderly population. Alzheimer's not only impacts individual cognitive decline but also places a significant psychosocial and economic burden on families and healthcare systems. According to data from Alzheimer’s Disease International (ADI), approximately 55 million people were living with dementia in 2020, and this number is projected to increase to 139 million by 2050 [1, 2]. Complementing these projections, recent epidemiological analyses have shown that the prevalence of Alzheimer’s disease (AD) and related dementias among individuals aged ≥65 years increased more than 1.6-fold between 1991 and 2021, reaching nearly 49 million cases in 2021 [3]. This marked increase underscores the progressive nature of the disease and its growing global burden on patients, caregivers, and healthcare systems [4].
Pathologically, Alzheimer's disease is characterized by the accumulation of β-amyloid (Aβ) plaques and neurofibrillary tangles (NFTs), which progressively damage neural tissue [5]. Oxidative stress has also been recognized as a key contributor to AD pathogenesis and is closely associated with amyloid beta accumulation, tau pathology, and neuronal degeneration [6]. Aβ plaques are formed through the processing of amyloid precursor protein (APP) by the BACE1 enzyme, which has become a key target for therapeutic development. Increased BACE1 activity is directly linked to the accumulation of neurotoxic Aβ and has also been associated with downstream pathological events such as tau hyper phosphorylation, neuroinflammation, and synaptic dysfunction [7-10]. BACE2 is a homolog of BACE1. They have similar structures, but their functions are different. BACE2 mainly works in other metabolic processes, such as proinsulin processing [11]. Therefore, developing inhibitors that selectively target BACE1 without affecting BACE2 is a critical challenge in Alzheimer's therapy.
Sulforaphane, an isothiocyanate compound derived from cruciferous vegetables, exhibits antioxidant and anti-inflammatory activities that contribute to its neuroprotective effects [12, 13]. Sulforaphane has been reported to non-competitively inhibit BACE1 with an IC₅₀ value of 2.08±0.19 µM and a Ki of 3.1 µM, showing approximately six-fold higher potency than resveratrol and quercetin, while exerting minimal effects on BACE2 and other proteases, thus highlighting its selectivity and potential as a safe natural BACE1 inhibitor without significant effects on BACE2 [14]. Given that sulforaphane functions as a non-competitive inhibitor, this research is dedicated to mapping its potential allosteric binding site within the BACE1 enzyme. Pinpointing the exact location and mechanism of this molecular interaction is highly relevant. This is due to the fact that allosteric inhibitors typically exhibit higher selectivity and often result in fewer adverse effects compared to their competitive counterparts. In this context, sulforaphane emerges as a promising candidate that could interact with BACE1 with greater specificity could pave the way for developing therapeutic options that are both safer and more accurately targeted.
Molecular docking and molecular dynamics simulations are powerful computational tools for investigating the interactions between small molecules and target proteins. Molecular docking predicts the preferred binding site and orientation of a ligand, while molecular dynamics simulations provide a dynamic view of complex stability and conformational changes under near-physiological conditions[15-18]. We aimed to elucidate the structural and dynamic basis of sulforaphane’s allosteric inhibition of BACE1 through an integrated computational approach. This strategy combined molecular docking to predict a putative allosteric binding site with molecular dynamics simulations to evaluate the stability and molecular interactions of the resulting complexes. These findings provide a structural framework for the rational design of selective allosteric BACE1 inhibitors and support the development of improved therapeutic strategies for Alzheimer’s disease.
MATERIALS AND METHODS
Equipments
The tools used in this study were Omen by HP 15-dc1xxx laptop powered by an Intel Core i7-9750H processor (2.60 GHz) and 16 GB of RAM. The operating systems used were Windows 11 and Ubuntu 24.04.1 lTS. Some of the software used in this study include YASARA Structure 25.1.13, PyPLIF (Python based Protein Ligand Interaction Fingerprinting) HIPPOS 0.2.0, and GROMACS 2024.4. In addition, this study also uses a Cloud Protein Simulator (CPS) server equipped with a Graphics Processing Unit (GPU) from molmodid for the molecular dynamics simulation process.
Input file preparation
The BACE1 structure used in this study was downloaded from the RCSB Protein Data Bank (PDB ID: 2WJO). The PDB structure download process was carried out directly within the YASARA Structure program. The next step was to identify and correct the missing amino acid residues. In this crystal structure, the missing residues were found in three regions: the loop, N-terminal, and C-terminal. The missing residues were then constructed using YASARA Structure. After model reconstruction, the protein was adjusted to near-physiological conditions by setting the system pH to 7.4, followed by energy minimization using the AMBER14 force field implemented in YASARA Structure. The co-crystallized native ligand was then removed, and the finalized model was stored as BACE1_receptor.pdb for subsequent analyses [19].
The three-dimensional structure of sulforaphane was constructed in YASARA-Structure using the Build molecule from SMILES string function, with the SMILES notation entered as CS(=O)CCCCN=C=S. The system was then adjusted to physiological conditions (pH 7.4) and subjected to energy minimization to achieve a stable conformation. The optimized ligand was subsequently saved as BACE1_ligand.Pdb [19].
Identification of allosteric site
To locate possible allosteric binding regions, molecular docking was first performed and followed by a clustering-based assessment of the resulting poses. In this step, the dock_run.mcr macro available in YASARA-Structure was employed, and the docking procedure was repeated 1000 times to capture a wide range of ligand orientations within the protein cavity. The ensemble of docking poses was then grouped using a 4.5 Å cutoff distance, a criterion that enabled identification of recurring conformations representing the most probable allosteric binding sites.
Redocking simulation of the ligand
Following the identification of the allosteric site, we performed an iterative redocking procedure to refine the ligand's binding pose within the BACE1 allosteric pocket. This refinement, conducted 100 times using YASARA Structure with the molmod plugin [20], aimed to determine the most favorable ligand conformation. We subsequently clustered the resulting poses using a 2 Å cutoff to identify the most representative binding mode. This optimal pose served as the initial coordinates for the subsequent molecular dynamics simulations.
Molecular dynamics simulations
We conducted molecular dynamics simulations using the GROMACS software suite on the Cloud Protein Simulator (CPS) provided by molmod.id, employing a dedicated graphics processing unit GPU for accelerated computation. The protein was modeled with the AMBER14 force field, and sulforaphane parameters were derived from the General AMBER Force Field (GAFF). This represents one commonly adopted and internally compatible parameterization approach for protein-ligand simulations in GROMACS, rather than a mandatory force-field choice. We solvated the system in a cubic TIP3P-FB water box with an 8 Å buffer between the solute and box edge, then neutralized it and added Na⁺ and Cl⁻ ions to a final physiological concentration of 0.15 M NaCl. The system underwent energy minimization via steepest descent and conjugate gradient algorithms, followed by stepwise equilibration: 200 ps under NVT and 500 ps under NPT ensembles at 310 K and 1 atm. For the production phase, we ran five independent 10ns simulations, saving coordinates every 10 ps. Complex stability was evaluated by calculating the Root mean Square Deviation of the protein backbone (RMSDBb) and the Root mean Square Deviation of Ligand Move (RMSDLigMove). In this analysis, the simulation trajectories were first aligned to the protein backbone, and RMSDLigMove was then calculated to isolate the true displacement of the ligand within the binding pocket, independent of the overall protein motion.
Interaction analysis
Analysis of the interactions formed during the molecular dynamics simulation process was carried out using PyPLIF HIPPOS. The analysis was performed over the last 5 ns of the simulation. The interaction analysis was conducted using the direct IFP method to identify interactions that occurred during the molecular dynamics process. The types of interactions analyzed using PyPLIF HIPPOS included hydrophobic and non-hydrophobic bonds [21, 22].
RESULTS
Identification of allosteric site and redocking simulation
We identified the allosteric binding site for sulforaphane on BACE1 by integrating molecular docking with a clustering analysis. Using YASARA Structure, we performed global docking simulations, allowing sulforaphane to sample the entire protein surface to locate potential binding pockets. A total of 1000 independent docking runs were conducted to comprehensively map the interaction landscapes. The resulting poses were ranked according to their calculated free energy of binding (feb), which provided the primary metric for the subsequent clustering analysis. The feb values for these top-ranked poses ranged from –4.2090 to –3.1270 kcal/mol.
The binding pocket corresponding to ligand conformation 0094 was selected from the global docking analysis, and the visualization of this conformation is shown in fig. 1. This conformation was subsequently used as the starting structure for the redocking process. The representative docking poses of the resulting clusters are presented in fig. 2.
Molecular dynamics simulations
Molecular dynamics simulations were carried out using GROMACS with a 10 ns production time, executed in five parallel replicates. The stability of the sulforaphane-BACE1 complexes was evaluated using RMSD-based metrics. Ligand positional stability was assessed using RMSDLigMove, while protein conformational stability was monitored using RMSDBb. The RMSDLigMove profiles obtained from the five independent simulations are shown in fig. 3, whereas the RMSDBb profiles for each replicate are presented in fig. 4.
The molecular dynamics simulations performed in this study were limited to the sulforaphane-BACE1 complex only. No apo-BACE1 or competitive inhibitor-bound reference simulations were included, and no dynamic communication analyses such as DCCM, RMSF, or radius of gyration (Rg) were performed. Accordingly, the observed RMSDLigMove and RMSDBb profiles describe ligand behavior in isolation and do not provide comparative evidence of ligand-induced stabilization or allosteric signal propagation. The results therefore reflect the intrinsic stability and variability of sulforaphane binding within the predicted allosteric pocket. Notably, the sulforaphane-BACE1 complex was not consistently stable across all replicas, as only two of five simulations maintained stable ligand positioning.
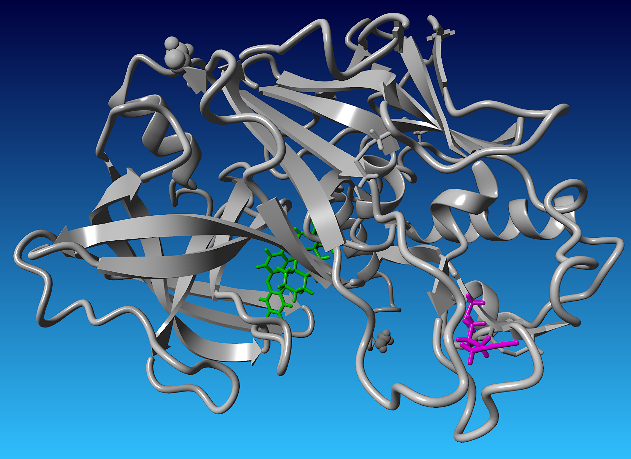
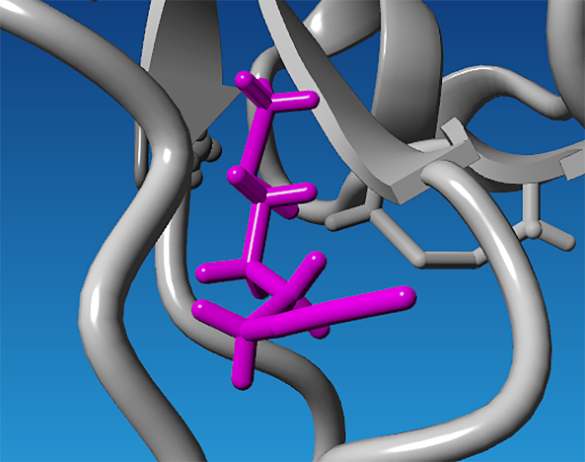
(a) (b)
Fig. 1: Identification of the sulforaphane allosteric binding site on BACE1 based on global docking and clustering analysis. (a) Surface representation of BACE1 showing sulforaphane bound to an allosteric site (magenta) that is spatially distinct from the catalytic dyad (green). (b) Zoomed-in view of the allosteric binding pocket illustrating the residues that define the binding environment. The spatial separation of this pocket from the active site suggests its potential relevance for allosteric modulation of BACE1 activity without direct competition with substrate binding
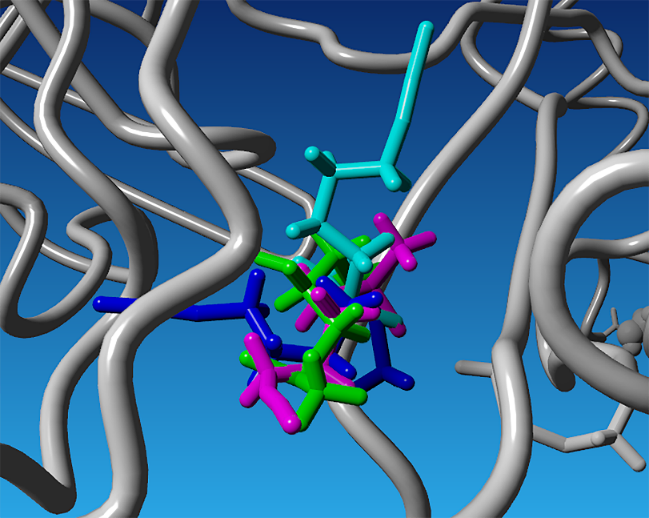
Fig. 2: Superimposed binding conformations of sulforaphane at the identified allosteric site of BACE1 obtained from redocking. The displayed poses represent four distinct ligand conformations corresponding to the main clusters identified after clustering analysis, shown in different colors (magenta, cyan, green, and blue)
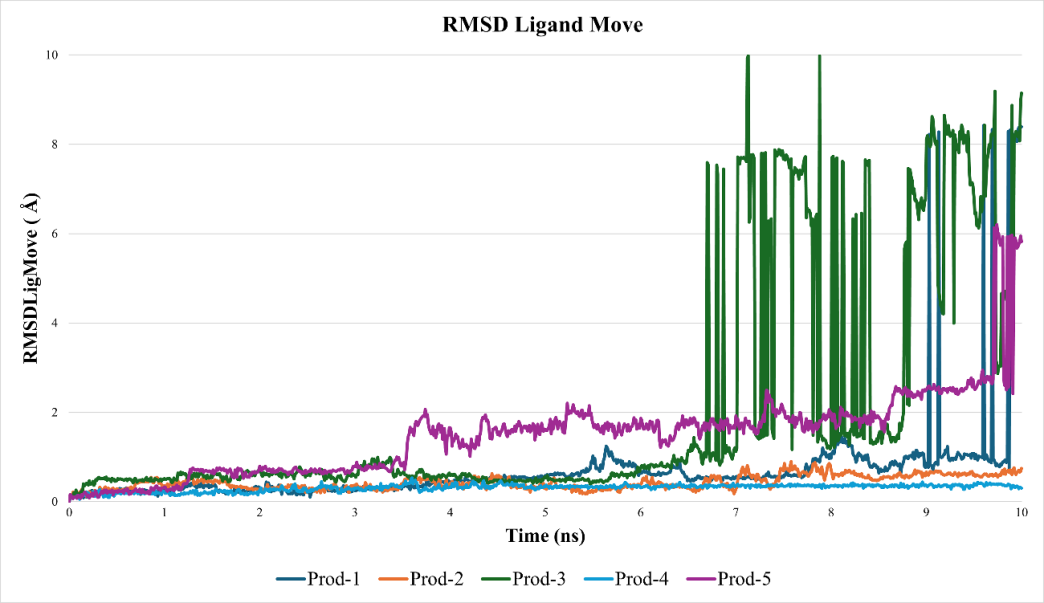
Fig. 3: RMSDLigMove profiles of sulforaphane during five independent molecular dynamics simulations. Ligand stability was observed in runs 2 and 4, whereas runs 1, 3, and 5 exhibited greater displacement, suggesting the presence of multiple metastable states. y-axis label: RMSDLigMove (Å)
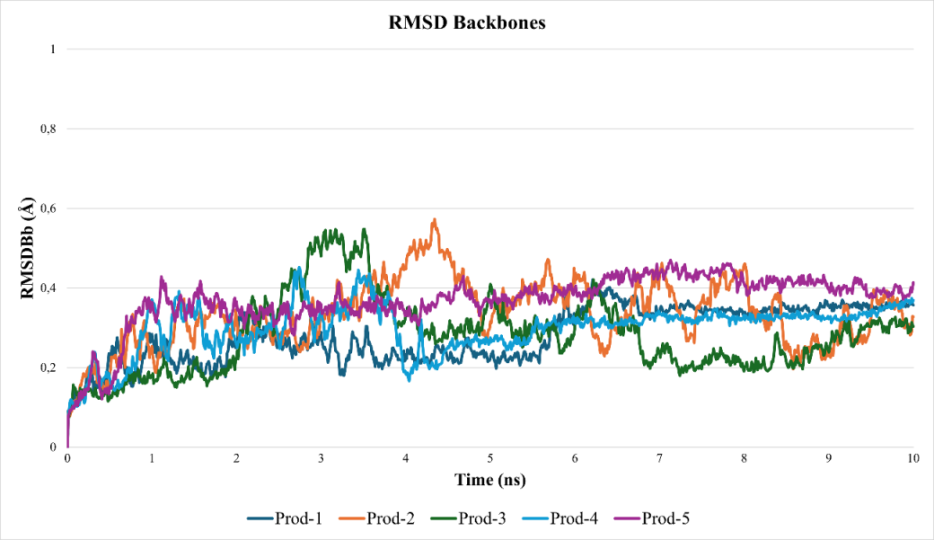
Fig. 4: RMSD profiles of the BACE1 protein backbone (RMSDBb) obtained from five independent molecular dynamics simulations. Low RMSD values (<2 Å) across all replicates confirm that the overall protein folding remained intact throughout the simulations, indicating that ligand movement was not associated with global protein unfolding
Interaction analysis
Interaction fingerprint analysis using PyPLIF HIPPOS 0.2.0 was performed to characterize the binding profile of sulforaphane at the allosteric site of BACE1 during molecular dynamics simulations. The hydrophobic and non-hydrophobic interactions between sulforaphane and BACE1 are presented in table 1 and table 2, respectively.
Table 1: Hydrophobic interaction hotspots in the sulforaphane–BACE1 complex identified by PyPLIF HIPPOS 0.2.0 during 5–10 ns
| Interacting residue | Interaction type | Interaction percentage (%) | ||||
| R1 | R2 | R3 | R4 | R5 | ||
| ALA162 | Hydrophobic | 65.47 | 2.59 | 3.39 | 3.79 | - |
| ALA173 | Hydrophobic | 28.74 | 52.30 | 23.95 | 95.21 | - |
| ALA255 | Hydrophobic | - | - | - | - | 0.40 |
| ALA256 | Hydrophobic | - | - | - | - | 3.39 |
| ALA277 | Hydrophobic | - | - | - | 1.40 | - |
| ALA318 | Hydrophobic | - | 3.79 | 34.93 | - | - |
| ASN167 | Hydrophobic | - | 3.79 | 18.16 | 41.72 | - |
| ASN214 | Hydrophobic | - | - | - | - | 18.16 |
| ASN283 | Hydrophobic | - | - | - | - | 4.99 |
| ASP316 | Hydrophobic | - | - | 3.19 | - | - |
| ASP323 | Hydrophobic | 1.40 | 31.14 | 1.40 | 60.88 | - |
| ASP368 | Hydrophobic | 22.55 | - | - | - | 58.68 |
| CYS324 | Hydrophobic | - | 4.79 | - | - | - |
| GLN168 | Hydrophobic | 5.99 | 17.96 | - | - | - |
| GLN308 | Hydrophobic | 31.74 | - | - | - | 36.93 |
| GLU315 | Hydrophobic | 9.98 | 33.53 | 23.95 | 78.64 | - |
| GLU369 | Hydrophobic | 1.00 | - | - | - | 63.27 |
| HIS367 | Hydrophobic | 0.20 | - | - | - | - |
| HIS54 | Hydrophobic | - | - | 0.80 | - | - |
| ILE284 | Hydrophobic | - | - | - | - | 0.20 |
| LEU166 | Hydrophobic | 24.55 | 78.44 | - | 3.39 | - |
| LEU172 | Hydrophobic | 31.14 | 12.97 | 36.53 | 93.41 | - |
| PHE164 | Hydrophobic | 5.19 | - | - | - | - |
| PHE370 | Hydrophobic | - | - | - | - | 32.53 |
| PRO165 | Hydrophobic | 1.00 | - | 13.37 | 10.38 | - |
| PRO307 | Hydrophobic | - | - | - | - | 0.80 |
| PRO313 | Hydrophobic | 51.50 | 82.44 | 16.77 | 99.60 | 0.20 |
| PRO49 | Hydrophobic | - | - | 0.80 | - | - |
| PRO51 | Hydrophobic | - | - | 0.40 | - | - |
| THR279 | Hydrophobic | - | - | - | - | 0.40 |
| THR304 | Hydrophobic | - | - | - | - | 0.20 |
| THR319 | Hydrophobic | - | - | 0.20 | - | - |
| TRP282 | Hydrophobic | 43.11 | 0.60 | - | - | 67.86 |
| TYR325 | Hydrophobic | 33.33 | 26.35 | - | 26.55 | - |
| TYR56 | Hydrophobic | - | - | 1.20 | - | - |
| VAL171 | Hydrophobic | 0.80 | - | - | - | - |
| VAL175 | Hydrophobic | 1.40 | 2.59 | 7.19 | - | - |
| VAL287 | Hydrophobic | - | - | - | - | 15.57 |
| VAL314 | Hydrophobic | - | 1.40 | - | 0.60 | - |
| VAL317 | Hydrophobic | - | - | 5.99 | - | - |
| VAL366 | Hydrophobic | 0.80 | 0.20 | - | 0.20 | - |
Notes: Five replicates of production runs were coded with R1, R2, R3, R4, and R5.
Table 2: Non-hydrophobic interaction hotspots in the sulforaphane–BACE1 complex identified by PyPLIF HIPPOS 0.2.0 during 5–10 ns
| Interacting residue | Interaction type | Interaction percentage (%) | ||||
| R1 | R2 | R3 | R4 | R5 | ||
| ARG312 | H-bond donor | - | - | - | 1.20 | - |
| ARG312 | Ionic as the cation | - | - | - | 0.80 | - |
| ARG371 | H-bond donor | - | - | - | - | 6.59 |
| ASN167 | H-bond donor | - | 0.60 | 5.79 | 2.20 | - |
| ASN214 | H-bond donor | - | - | - | - | 0.80 |
| ASN283 | H-bond donor | - | - | - | - | 2.99 |
| ASP323 | H-bond acceptor | - | 0.40 | - | - | - |
| ASP323 | Ionic as the anion | - | 4.99 | - | 1.20 | - |
| ASP368 | Ionic as the anion | 0.40 | - | - | - | 5.39 |
| GLN17 | H-bond donor | - | 2.00 | - | - | - |
| GLN308 | H-bond donor | 0.20 | - | - | - | 0.80 |
| GLU315 | H-bond acceptor | - | - | 0.20 | - | - |
| GLU315 | Ionic as the anion | - | 0.20 | 5.79 | - | - |
| GLU369 | Ionic as the anion | - | - | - | - | 0.40 |
| SER320 | H-bond donor | - | 1.00 | 0.40 | - | - |
| TYR325 | H-bond acceptor | 2.00 | 0.60 | - | - | - |
| TYR325 | H-bond donor | - | 0.20 | - | - | - |
Notes: Five replicates of production runs were coded with R1, R2, R3, R4, and R5.
DISCUSSION
The molecular docking results provide insight into the binding energetics of sulforaphane at the predicted allosteric site of BACE1. The calculated Free Energy of Binding (feb) values ranged from −4.2 to −3.9 kcal/mol, indicating weak but specific interactions between sulforaphane and the predicted binding pocket. These values suggest a moderate binding affinity, which is characteristic of ligand s targeting flexible, non-catalytic regulatory sites rather than deeply buried active sites. The docking poses revealed that sulforaphane engages the allosteric region primarily through hydrophobic interactions and limited polar contacts, supporting its role as a modulator rather than a tight-binding inhibitor.
Critically, the feb values obtained from the docking simulations (–4.2 to –3.9 kcal/mol) are qualitatively consistent with the experimental kinetic data reported by [14], which describe sulforaphane as a non-competitive BACE1 inhibitor with a Ki of 3.1 µM. This qualitative agreement supports the plausibility of the predicted binding modes, without implying quantitative validation of inhibitory potency or binding affinity. Taken together, these docking results provide a structural context for sulforaphane’s moderate inhibitory activity and serve as a basis for interpreting its dynamic behavior in the subsequent molecular dynamics simulations.
We identified the predominant binding pockets through a clustering analysis of the docking results, grouping ligand poses by the spatial similarity of their binding locations [23]. This analysis employed a 4.5 Å distance cut off a common threshold for capturing key hydrogen bonding and van der Waals interactions involved in ligand recognition and stabilizationto define associated poses and pocket-forming residues [24]. By sequentially assigning docking conformations to clusters based on their feb values, we identified 35 distinct clusters that represent the major binding regions sampled by sulforaphane on the BACE1 surface.
The next step involved visually grouping the identified binding pockets based on the position of the sulforaphane ligand within the BACE1 structure. This classification was divided into three categories: (i) ligand located inside the protein but outside the catalytic site, (ii) ligand positioned near the protein surface but still outside the catalytic site, and (iii) ligand directly bound to the catalytic site. Based on this classification, the first category where the ligand binds within the protein but outside the catalytic sitewas prioritized for further analysis. Binding pockets belonging to this category were then examined visually to identify the most promising candidates. From these observations, the binding pocket with ligand conformation 0094 was selected from the global docking results, which had the best position. In addition, this conformation also showed the lowest feb value.
In the following stage, the sulforaphane ligand was redocked into the selected binding pocket. The redocking procedure was repeated 100 times, resulting in binding energy values ranging between –4.3530 and –3.9240 kcal/mol. The generated poses were subjected to clustering based on structural similarity, applying a 2 Å distance cutoff [23]. Based on binding energy as the reference criterion, four clusters were obtained: cluster 1 (92 poses), cluster 2 (5 poses), cluster 3 (2 poses), and cluster 4 (1 pose). The representative docking poses of these clusters are shown in fig. 2. Among them, cluster 1 was selected as the most representative for subsequent molecular dynamics simulations.
The stability of the sulforaphane–BACE1 complex through five independent 10 ns molecular dynamics simulations. Following previous studies on ligand binding to flexible sites [25, 26], an RMSDLigMove value below 2 Å was considered indicative of a stable and productive binding mode. The analyses revealed replica-dependent variability (fig. 3). Two trajectories (R2 and R4) maintained consistent ligand positioning within the pocket, with final RMSDLigMove values of 0.725 Å and 0.173 Å, respectively. In contrast, the remaining simulations exhibited larger fluctuations, with ligand displacement reaching approximately 9.6 Å, suggesting reorientation or transient dissociation events. Despite these differences, the protein backbone remained stable across all replicates, with RMSDBb values below 2 Å (fig. 4). This pattern indicates that sulforaphane can adopt a stable binding orientation in specific trajectories (R2 and R4), whereas others reflect alternative or metastable states, consistent with moderate affinity and the intrinsic flexibility of allosteric regions. Accordingly, the sulforaphane-BACE1 complex should not be interpreted as uniformly stable across all simulations, but rather as exhibiting replica-dependent and context-specific binding behavior.
In the present study, the molecular dynamics analysis was intentionally designed as a preliminary and non-comparative evaluation of the structural stability and binding feasibility of the sulforaphane-BACE1 complex. The simulation framework focused on assessing whether sulforaphane can adopt and maintain a stable orientation within a predicted allosteric pocket using RMSD-based stability parameters. Because no apo-BACE1 or competitive inhibitor-bound reference systems were simulated, and dynamic communication analyses such as DCCM, per-residue RMSF, and Rg were not performed, the present results do not allow direct assessment of ligand-induced stabilization, catalytic dyad modulation, or long-range allosteric signal propagation. The observed RMSDLigMove behavior and interaction persistence therefore describe the intrinsic dynamics of the ligand-bound system only. While comparative simulations and advanced dynamic analyses would be required to establish mechanistic allosteric regulation and communication between the allosteric site and the catalytic dyad (ASP32, ASP228), such investigations were beyond the scope of this initial study. These analyses are identified as essential future work to extend the present computational framework and to substantiate the proposed allosteric inhibition mechanism.
Selectivity for BACE1 over its homolog BACE2 is a crucial pharmacological consideration, as off-target inhibition of BACE2 has been associated with adverse effects. It should be noted that the present study did not include docking or molecular dynamics simulations against BACE2, and therefore does not provide computational or structural evidence for BACE1–BACE2 selectivity. The discussion of selectivity in this work is instead based on previously reported experimental data. Enzymatic assays have shown that sulforaphane potently inhibits BACE1 activity (IC₅₀ = 2.08±0.19 µM), while exhibiting minimal inhibition of BACE2, with only 6–11% activity reduction even at concentrations 25–50 times higher (50–100 µM) [14]. These results indicate a clear functional preference for BACE1 under experimental conditions.
The same study also reported a non-competitive inhibition mechanism for sulforaphane, suggesting binding to an allosteric site distinct from the highly conserved catalytic domain [14]. Because allosteric pockets often display greater structural divergence among homologous enzymes, this mechanism provides a plausible explanation for the observed experimental selectivity. In this context, the present computational model suggests that sulforaphane can form a moderately stable complex with a predicted allosteric site on BACE1. However, this model does not mechanistically confirm selectivity over BACE2. Comparative docking and molecular dynamics simulations involving BACE2 will therefore be required to directly address this question and are identified as essential future work.
Interaction fingerprint analysis was conducted to elucidate the molecular determinants responsible for the differential stability observed across the simulations. The refined analysis focused on persistent contacts (>50%) identified in the stable trajectories (R2 and R4). The results revealed that hydrophobic interactions dominate the stabilization of the sulforaphane–BACE1 complex, with ALA173, LEU172, PRO313, GLU315, and ASP323 consistently maintaining high contact frequency throughout these simulations. Among them, ASP323 emerged as a principal anchoring residue, forming both hydrophobic and electrostatic interactions that secure the ligand orientation within the allosteric pocket.
The interaction network in R2 and R4 showed recurrent and convergent binding patterns, suggesting a well-defined productive pose, whereas other replicas displayed less persistent or transient contacts. This replica-dependent variability reflects the inherent flexibility of the BACE1 allosteric region, which accommodates multiple metastable states. Overall, the persistence of hydrophobic and auxiliary polar contacts, particularly those centered around ASP323, supports a mechanistic model in which sulforaphane achieves moderate but reproducible stabilization within the allosteric pocket of BACE1.
CONCLUSION
This study presents a preliminary and non-comparative computational evaluation of sulforaphane binding at a putative allosteric site of BACE1. The molecular dynamics simulations were limited to the ligand-bound system, without apo or reference inhibitor-bound controls and without dynamic communication analyses. Accordingly, the findings describe the feasibility and replica-dependent stability of sulforaphane binding within a flexible allosteric region, rather than definitive ligand-induced stabilization or mechanistic confirmation of allosteric inhibition.
Stable binding conformations observed in selected trajectories (R2 and R4) were primarily supported by hydrophobic interactions involving ALA173, LEU172, PRO313, GLU315, and the anchoring residue ASP323, with additional transient polar contacts. Importantly, the computational model presented here should be interpreted as a hypothesis-generating structural framework to support future comparative simulations and rational design of sulforaphane analogs, rather than as confirmation of inhibitory potency.
ACKNOWLEDGEMENT
The authors express their sincere gratitude to molmod. id for providing mentorship and access to computational resources throughout this study. This research was financially supported by the DPPM program of the Government of the Republic of Indonesia with contract number 126/C3/DT.05.00/PL/2025 awarded to Dr. Florentinus Dika Octa Riswanto.
AUTHORS CONTRIBUTIONS
M. O conducted all the computational study, data analysis, and initial manuscript writing. M. H. P. performed the protocol development and manuscript review. B. I. W. carried out the computational supervision and manuscript review. E. P. I. conceptualized the research project and supervised the computational study. F. D. O. R conceptualized the research project, provided the research funding, and performed the correspondence of the publication.
CONFLICT OF INTERESTS
The authors declare the following competing financial interest(s): E. P. I. is a co-founder and B. I. W. is a Research and General Affairs Officer of Molmod Jaya Sejahtera, Ltd. (MOLMOD ID), a contract research organization that provides fee-for-service cloud protein simulators and molecular modeling simulations. The authors declare no other conflicts.
REFERENCES
Nichols E, Steinmetz JD, Vollset SE, Fukutaki K, Chalek J, Allah FA. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the global burden of disease study 2019. Lancet Public Health. 2022;7(2):e105-25. doi: 10.1016/S2468-2667(21)00249-8.
Xiaopeng Z, Jing Y, Xia L, Xingsheng W, Juan D, Yan L. Global burden of Alzheimer’s disease and other dementias in adults aged 65 y and older, 1991-2021: population-based study. Front Public Health. 2025;13:1585711. doi: 10.3389/fpubh.2025.1585711, PMID 40666154.
Sakshi K, Mote S, Supekar A, Saudar S. A review on antibody of aducanumab- reduces the progression of Alzheimer disease. Int J Pharm Pharm Sci. 2023;5(1):15-22. doi: 10.33545/26647222.2023.v5.i1a.28.
De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164(4):603-15. doi: 10.1016/j.cell.2015.12.056, PMID 26871627.
Swetha G, Raj A, Tabassum S, Chhakchhuak DZ. Oxidative stress in Alzheimer’s disease-evaluating the amyloid beta hypothesis. Int J Curr Pharm Sci. 2021;13(5):32-8. doi: 10.22159/ijcpr.2021v13i5.1906.
Vassar R. BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease. Alzheimers Res Ther. 2014;6(9):89. doi: 10.1186/s13195-014-0089-7, PMID 25621019.
Hampel H, Vassar R, De Strooper BD, Hardy J, Willem M, Singh N. The β-secretase BACE1 in Alzheimer’s disease. Biol Psychiatry. 2021;89(8):745-56. doi: 10.1016/j.biopsych.2020.02.001, PMID 32223911.
Abubakar MB, Sanusi KO, Ugusman A, Mohamed W, Kamal H, Ibrahim NH. Alzheimer’s disease: an update and insights into pathophysiology. Front Aging Neurosci. 2022;14:742408. doi: 10.3389/fnagi.2022.742408, PMID 35431894.
Kamatham PT, Shukla R, Khatri DK, Vora LK. Pathogenesis diagnostics and therapeutics for Alzheimer’s disease: breaking the memory barrier. Ageing Res Rev. 2024;101:102481. doi: 10.1016/j.arr.2024.102481, PMID 39236855.
Fujimoto K, Matsuoka E, Asada N, Tadano G, Yamamoto T, Nakahara K. Structure-based design of selective β-Site amyloid precursor protein cleaving enzyme 1 (BACE1) inhibitors: targeting the flap to gain selectivity over BACE2. J Med Chem. 2019;62(10):5080-95. doi: 10.1021/acs.jmedchem.9b00309, PMID 31021626.
Schepici G, Bramanti P, Mazzon E. Efficacy of sulforaphane in neurodegenerative diseases. Int J Mol Sci. 2020;21(22):8637. doi: 10.3390/ijms21228637, PMID 33207780.
Zhang Y, LV C, Sun J, Song X, Makaza N, Wu Y. Protective effects of broccoli extracts and sulforaphane against hydrogen peroxide induced oxidative stress in B16 cells. J Funct Foods. 2021;87:104833. doi: 10.1016/j.jff.2021.104833.
Youn K, Yoon JH, Lee N, Lim G, Lee J, Sang S. Discovery of sulforaphane as a potent BACE1 inhibitor based on kinetics and computational studies. Nutrients. 2020;12(10):3026. doi: 10.3390/nu12103026, PMID 33023225.
Shakil S, Rizvi SM, Greig NH. High throughput virtual screening and molecular dynamics simulation for identifying a putative inhibitor of bacterial CTX-M-15. Antibiotics (Basel). 2021;10(5):474. doi: 10.3390/antibiotics10050474, PMID 33919115.
Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018;99(6):1129-43. doi: 10.1016/j.neuron.2018.08.011, PMID 30236283.
Agu PC, Afiukwa CA, Orji OU, Ezeh EM, Ofoke IH, Ogbu CO. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci Rep. 2023;13(1):13398. doi: 10.1038/s41598-023-40160-2, PMID 37592012.
Dnyandev KM, Babasaheb GV, Chandrashekhar KV, Chandrakant MA, Vasant OK. A review on molecular docking. IRJPAC. 2021;22(3):60-8. doi: 10.9734/irjpac/2021/v22i330396.
Istyastono EP, Octa Riswanto FD. Molecular dynamics simulations of the caffeic acid interactions to dipeptidyl peptidase IV. Int J App Pharm. 2022;14(4):274-8. doi: 10.22159/ijap.2022v14i4.44631.
Istyastono EP. Simulasi dan validasi penambatan molekul dengan pugasan YASARA-structure. Yogyakarta: Sanata Dharma University Press; 2023.
Wiranata BI, Istyastono EP. Interaction dynamics of caffeine in the human acetylcholinesterase binding pocket. J Food Pharm Sci. 2025;13(1):1-9. doi: 10.22146/jfps.16533.
Istyastono EP, Yuniarti N, Prasasty VD, Mungkasi S, Waskitha SS, Yanuar MR. Caffeic acid in spent coffee grounds as a dual inhibitor for MMP-9 and DPP-4 enzymes. Molecules. 2023;28(20):7182. doi: 10.3390/molecules28207182, PMID 37894660.
Makeneni S, Thieker DF, Woods RJ. Applying pose clustering and md simulations to eliminate false positives in molecular docking. J Chem Inf Model. 2018;58(3):605-14. doi: 10.1021/acs.jcim.7b00588, PMID 29431438.
Clark JJ, Orban ZJ, Carlson HA. Predicting binding sites from unbound versus bound protein structures. Sci Rep. 2020;10(1):15856. doi: 10.1038/s41598-020-72906-7, PMID 32985584.
Liu K, Watanabe E, Kokubo H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J Comput Aid Mol Des. 2017;31(2):201-11. doi: 10.1007/s10822-016-0005-2, PMID 28074360.
Liu K, Kokubo H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations: a cross-docking study. J Chem Inf Model. 2017;57(10):2514-22. doi: 10.1021/acs.jcim.7b00412, PMID 28902511.