
Int J Curr Pharm Res, Vol 17, Issue 4, 26-31Review Article
SELF MICROEMULSIFYING DRUG DELIVERY SYSTEMS (SMEDDS): KEY APPROACH FOR IMPROVING ORAL DELIVERY OF POORLY WATER SOLUBLE DRUGS
DINAKARAN M., GRACE RATHNAM*
Department of Pharmaceutics, C. L. Baid Metha College of Pharmacy, Rajiv Gandhi Salai, Thoraipakkam, Chennai-600097, India
*Corresponding author: Grace Rathnam; *Email: gracerathnam@clbaidmethacollege.com
Received: 18 Apr 2025, Revised and Accepted: 05 Jun 2025
ABSTRACT
Nearly 50% of newly developed drug candidates that progress to the formulation stage exhibit poor water solubility. Oral administration remains the most common route for the long-term treatment of many diseases. However, in numerous cases, the orally delivered drugs encounter obstacles due to their high lipophilicity. These poorly soluble in water face challenges such as reduced oral bioavailability, significant variability between and within subjects, and a lack of dose proportionality. The primary goal of this review is to compile and present detailed information on the design and assessment of self microemulsifying drug delivery systems (SMEDDS). This compilation aims to assist in improving the bioavailability of poorly water-soluble drugs administered orally. SMEDDS are isotropic mixtures of oils, surfactants, and co-surfactants. Upon mild agitation and dilution with water-such as gastrointestinal fluids-they form clear oil-in-water microemulsions. SMEDDS has become a key strategy for enhancing the bioavailability of poorly water-soluble drugs. Despite its benefits, SMEDDS faces several challenges, such as drug precipitation in the body, handling difficulties, limited uptake through the lymphatic system, the absence of reliable in vitro predictive tests, and the risk of oxidation of unsaturated fatty acids. These drawbacks can hinder its broader application. Converting SMEDDS into solid forms can also resolve issues related to the instability and handling of liquid formulations. This review thoroughly examines the challenges of SMEDDS and explores practical strategies to address them.
Keywords: Self-micro emulsifying drug delivery systems (SMEDDS), Oral bioavailability, Lipid phase, Surfactant, Co-surfactant
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijcpr.2025v17i4.7034 Journal homepage: https://innovareacademics.in/journals/index.php/ijcpr
INTRODUCTION
Self-micro emulsifying drug delivery systems (SMEDDS) are isotropic blends composed of natural or synthetic oils, solid or liquid surfactants, and one or more hydrophilic solvents and cosolvents/surfactants. These formulations have a distinctive ability to spontaneously form fine oil-in-water (o/w) microemulsions upon gentle agitation and subsequent dilution in aqueous environments, such as gastrointestinal (GI) fluids. When administered, SMEDDS disperse quickly within the GI tract, where the natural motility of the stomach and intestines provides the necessary agitation for self-emulsification [1].
The key difference between self-emulsifying drug delivery systems (SEDDS), also referred to as self-emulsifying oil formulations, and SMEDDS lies in their droplet size and composition. SEDDS typically generate opaque emulsions with droplet sizes ranging from 100 to 300 nm, whereas SMEDDS produce transparent microemulsions with droplet sizes below 50 nm. Additionally, SMEDDS contain less than 20% oil, whereas SEDDS formulations include between 40% and 80% oil [2].
Although various techniques can be employed to enhance the solubility of poorly water-soluble drugs and improve their bioavailability, SMEDDS are particularly advantageous due to their physical stability and ease of manufacturing. These systems are especially beneficial for lipophilic drugs with dissolution rate-limited absorption, as they can enhance both the rate and extent of drug absorption, leading to more consistent blood concentration profiles [3].
The oral route is the simplest and most convenient method for non-invasive drug administration. Due to its cost-effectiveness, oral drug delivery systems have consistently dominated the global drug delivery market. However, this route poses challenges for drug molecules with poor aqueous solubility. For a drug to be absorbed after oral administration, it must first dissolve in gastrointestinal (GI) fluids before permeating through biological membranes. Nearly 40% of newly developed chemical entities have low aqueous solubility, making their absorption a significant challenge in modern drug delivery. The primary factor limiting the absorption of these drugs is their dissolution in the GI tract. According to the Biopharmaceutical Classification System (BCS), such drugs fall under Class II, characterized by low solubility but high permeability. Various formulation strategies, including micronization, solid dispersion, and complexation with cyclodextrins, have been explored to enhance solubility. While these approaches have shown success in some cases, they also present certain limitations.
Since poorly water-soluble drugs are hydrophobic and highly lipophilic, lipid-based drug delivery systems are considered ideal for their formulation. SMEDDS are generally developed in liquid dosage forms and are often administered using soft gelatin capsules. However, these capsules present certain challenges, such as difficulties in manufacturing and compatibility issues with the gelatin shell. To address these drawbacks, solid-SMEDDS have been introduced, offering greater commercial potential and improved patient compliance. Several techniques have been developed to convert liquid SMEDDS into solid forms, including adsorption onto solid carriers, spray drying, spray cooling, melt extrusion, nanoparticle technology, and supercritical fluid-based processes.
Studies suggest that SMEDDS not only enhance drug absorption in the gastrointestinal (GI) tract but may also improve absorption through rectal and vaginal routes for poorly water-soluble drugs. Research has found that SMEDDS formulations containing long-chain triglycerides resulted in better drug absorption compared to those using medium-chain triglycerides [4]. SMEDDS offer multiple advantages, such as spontaneous self-emulsification, thermodynamic stability, increased bioavailability, and ease of formulation. Their ability to enhance solubility and drug absorption makes them a valuable approach in advanced drug delivery systems.
Need for SMEDDS
One approach to enhancing the oral delivery of poorly water-soluble compounds is to pre-dissolve the drug in a suitable solvent and encapsulate the formulation. The primary advantage of this method is that it eliminates the initial rate-limiting step of drug particle dissolution in the aqueous environment of the gastrointestinal tract (GIT) [5]. If a drug can be dissolved in a lipid-based system, the likelihood of precipitation upon dilution in the GIT is reduced, as partitioning kinetics favor the drug remaining within lipid droplets. Another technique for improving solubility involves formulating the drug as a solid solution using a water-soluble polymer. This helps enhance drug solubility; however, a major drawback is the tendency of the drug to transition into a more thermodynamically stable state, potentially leading to crystallization within the polymer matrix. Considering these challenges, SMEDDS offer an effective solution by ensuring the drug remains in a solubilized state, thereby improving its dissolution, absorption, and overall bioavailability [6-8].
Advantages of SMEDDS
Enhances oral bioavailability by improving drug solubility and facilitating efficient transport [9, 10].
Simplifies manufacturing and scale-up compared to other lipid-based dosage forms.
Minimizes variations in drug absorption between individuals and reduces the impact of food intake [11].
Enables the delivery of peptide-based drugs that are susceptible to enzymatic degradation in the gastrointestinal tract (GIT) [12].
Unlike other lipid-based drug delivery systems, SMEDDS are not affected by lipid digestion processes [13].
Incorporating polymers into SMEDDS formulations can enable sustained drug release [14-17].
Disadvantages of SMEDDS
There is a lack of reliable in vitro models for accurately assessing SMEDDS formulations [18].
Existing in vitro models require further refinement and validation to establish their predictive reliability.
The development of SMEDDS formulations relies on establishing in vitro-in vivo correlations, necessitating extensive testing in appropriate animal models [19].
Potential chemical instability of drugs and the high surfactant content (typically 30-60%) may cause gastrointestinal irritation.
Conventional self-microemulsifying formulations containing volatile co-solvents may migrate into the shells of soft or hard gelatin capsules, leading to drug precipitation [20].
Upon dilution, the hydrophilic solvent effect may increase the risk of drug precipitation, impacting formulation stability.
Composition of SMEDDS
Oil (Lipid phase)
Role
Acts as a solvent for the lipophilic drug, facilitates drug dissolution, and forms the oil core of the microemulsion. The oil phase also promotes lymphatic transport, bypassing hepatic first-pass metabolism.
Characteristics
Typically, non-polar lipids with good drug solubility; medium-chain triglycerides (MCTs) or long-chain triglycerides (LCTs) are preferred for their stability and emulsification properties [21].
Examples
Medium-chain triglycerides: Capmul MCM, Miglyol 812, Labrafac Lipophile WL 1349
Long-chain triglycerides: Olive oil, soybean oil, sesame oil
Modified oils: Labrafac CM10, Maisine 35-
Concentration: Typically 10–30% w/w, depending on the drug’s solubility and emulsification requirements.
Surfactant
Role
Reduces interfacial tension between oil and water, enabling spontaneous microemulsion formation. Surfactants stabilize the emulsion and enhance drug solubilization.
Characteristics
Non-ionic surfactants with high Hydrophilic-Lipophilic Balance (HLB) values (12–18) are preferred for safety and effective emulsification [22].
Examples
Tween 80 (Polysorbate 80), Cremophor EL (Polyoxyl 35 castor oil) Labra sol, Kolliphor HS 15
Concentration
Typically 30–60% w/w, as higher concentrations ensure fine droplet size and stability, but must balance toxicity concerns.
Co-surfactant (Co-solvent)
Role
Enhances the emulsification process by further reducing interfacial tension and improving the flexibility of the surfactant film. It also aids in solubilizing the drug and preventing precipitation in the gastrointestinal tract.
Characteristics
Short-to medium-chain alcohols or polar solvents that integrate into the surfactant layer to optimize microemulsion formation [23].
Examples
Polyethylene glycol (PEG) 400Propylene glycol, Transcutol HP (Diethylene glycol monoethyl ether), Ethanol
Concentration
Typically 10–30% w/w, adjusted to achieve optimal phase behaviour and drug solubility.
Drug (Active pharmaceutical ingredient)
Role
The therapeutic agent incorporated into the SMEDDS, typically a poorly water-soluble drug (BCS Class II or IV) that benefits from enhanced solubility and bioavailability [24].
Examples
Fenofibrate, methotrexate, telmisartan, 3,3'-diindolylmethane (DIM), carbamazepine.
Considerations
The drug must have sufficient solubility in the oil, surfactant, or co-surfactant mixture to ensure effective loading and stability.
Optional components (for solid SMEDDS)
Solid Carriers/Adsorbents
Used to convert liquid SMEDDS into solid SMEDDS (S-SMEDDS) for improved stability and ease of administration (e. g., in capsules or tablets) [25].
Examples: Calcium silicate, Neusilin US2, Aerosil 200
Limitations of SMEDDS
Although SMEDDS formulation has several advantages, there are certain limitations associated with this system represented in fig. 1.
Drug precipitation on dilution
Diluted SMEDDS undergo precipitation of drug in gastrointestinal fluid. A common requirement for the lipid formulations is that they should be able to keep the drug in the solubilized form in the gastrointestinal tract (GIT). Precipitation of the drug from the system nullifies the advantage offered by the lipid-based formulation system [26, 27].
The precipitation tendency of the drug on dilution is higher due to the dilution effect of the hydrophilic solvent. It thereby requires incorporation of polymers to minimize drug precipitation in vivo [28].
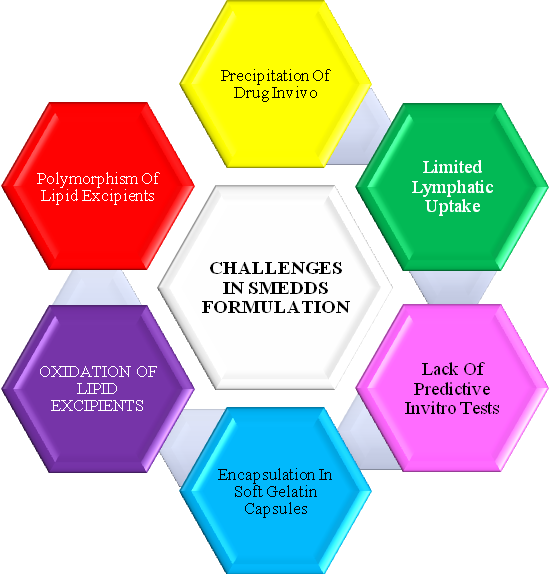
Fig. 1: Challenges in SMEDDS formulations
Encapsulation in soft gelatin capsules
Most of the marketed SMEDDS formulations are available as soft gelatin capsule. However, gelatin capsules are associated with few drawbacks. Manufacturing cost, transmissible spongiform encephalopathy (TSE) and consumer prefer-Ence/religion are the few issues associated with animal gelatin. Volatile co-solvents in self-microemulsifying formulations are known to migrate into the shells of soft or hard gelatin capsules, resulting in the precipitation of the lipophilic drugs. These problems drive the market requirement to find substitute for soft gelatin capsules. The current alternative material of choice to animal gelatin capsules are those prepared from HPMC. The HPMC capsule shell has been explored as an alternative approach for encapsulating super-saturable SMEDDS formulation [29].
Storage and handling
Liquid SMEDDS exhibit problems in handling, storage and stability. Thus, formulating solid SMEDDS seems to be a logical solution to address these problems [29].
Limited targeting to lymphatics
Targeting lymphatics confers two primary advantages over conventional absorption via the portal blood. First, transport through the intestinal lymph avoids pre-systemic hepatic metabolism and thereby enhances the concentration of orally administered drugs reaching the systemic circulation. Second, site-specific drug delivery to lymphatic organs could be achieved. Normally, high triglyceride solubility and high log P is required for lymphatic transport (Caliph et al.,) [30]. However, the amount of drug transported into lymphatics is variable from drug to drug. Hence, lipophilicity and triglyceride solubility of drug in correlation with lymphatic transport needs to be completely understood and a more adequate predictive model is required [31].
Lack of good in vitro models
Another obstacle in the development of SMEDDS and other lipid-based formulations is the lack of good predicative in vitro models for the assessment of the formulations [32]. Traditional dissolution methods do not work, as these formulations potentially are dependent on digestion of lipid in the gut, prior to release of the drug. Although to mimic this, an in vitro model simulating the digestive processes of the duodenum has been developed [33]. This in vitro model needs further refinement and validation before its strength can be evaluated. Further, development can be based on in vitro–in vivo correlations and therefore, different prototype lipid-based formulations need to be developed and evaluated in vivo in a suitable animal model [34-35].
Oxidation and polymorphism of the lipids used in formulating SEDDS/SMEDDS
Lipid excipients containing unsaturated fatty acids and its derivatives are prone to lipid oxidation [36]. This requires inclusion of lipid-soluble antioxidant in capsule formulation [37]. Polymorphism associated with thermo-softening lipid excipients requires specific process control in their application, in order to minimize polymorphic changes of the excipient matrix [38].
Handling issues of liquid smedds
SMEDDS are usually administered in the form of hard or soft gelatin capsules, with viscous liquids inside the lipid formulation into and interaction with the capsule shells is a possibility. This may lead to capsule shell softness or brittleness, leakage of the content, and drug or excipient creating a solid SMEDDS merge the advantages of solid materials with advantages of SMEDDS, e. g., increased solubility and bioavailability. Thus, the dosage forms (e. g., enhanced patient compliance, increased stability and reproducibility, simplicity of process control, low cost of manufacturing) [39-40]. Correspondingly, formulating liquid SMEDDS in solid dosage form produces a more consistent and stable form at a lower cost of manufacturing [41].
Dosing forms for solid SMEDDS
Dry emulsions
Dry emulsions are solid dosage forms that are in powder form and emulsify spontaneously when water is introduced. Spray drying, freeze drying, and rotary evaporation are ways of preparing dry emulsions. Spray drying is more frequently used when preparing dry emulsions for making dry emulsions. The o/w emulsion is made and then spray-dried to remove the aqueous phase [42].
Capsules
Solid SMEDDS can be filled into capsule shells using a range of technologies, such as spray drying and solid carrier adsorption. This prevents physical incompatibility. Liquid SMEDDS's ease of interaction with the capsule shell [43].
Tablets
Various liquid SMEDDS ratios may be adsorbed onto porous carriers. Other suitable excipients are then blended with the surface-adsorbed liquid SMEDDS. A compression machine is then employed to compress the blend. The irreversible precipitation of the drug inside the formulation was avoided by employing eutectic-based self-emulsifying tablets.
Encapsulation of liquid and semi-solid SMEDDS
It is simple to fill capsules with liquid SMEDDS and seal them using a banding technique or micro spray. The process of encapsulation of a semisolid SMEDDS involves four steps:
(1) Heating the semisolid excipients to a minimum of 20 ℃ above the melting point;
(2) Incorporating the drug while stirring in the molten blend;
(3) Filling the capsule shell with the drug-loaded molten blend; and
(4) Cooling the product to room temperature. SMEDDS encapsulation in capsules allows for high drug loading and is suitable for low-dose, very potent drugs [44, 45]
Spray drying
It is a widely adopted method for transforming liquid self-microemulsifying drug delivery systems (SMEDDS) into solid formulations. In this technique, the liquid SMEDDS is first mixed with a solid carrier dissolved in a suitable solvent. The mixture is then atomized into fine droplets and introduced into a drying chamber, where controlled temperature and airflow conditions promote rapid solvent evaporation. This process results in the formation of dry, free-flowing particles, enhancing the stability and handling of SMEDDS in solid dosage forms.
Adsorption on solid carriers
This involves incorporating the liquid formulation into porous powders or bead-like substances that possess a large surface area, capable of absorbing significant amounts of lipid material. According to Ito et al. [46], certain adsorbents can retain up to 70% of their own weight in liquid SMEDDS, making them highly efficient for this purpose. Materials such as Florite® and various grades of Neusilin®—including Neusilin US2 and UFL2-are commonly used due to their excellent oil-adsorbing capacities. This method not only stabilizes the formulation but also improves powder flow, making it suitable for subsequent processing into tablets or capsules [47].
Techniques for solidification in transforming liquid or semi-solid SMEDDS into SMEDDS
Spray drying
In this process essentially involves formulating a product by mixing lipids, solid carriers, drugs, and surfactants, and then dissolving the mixture before spray drying it. The dissolved liquid composition is then atomized to produce a droplet spray. The droplets are dried in a drying chamber with controlled airflow and temperature, where the volatile phase (e. g., water of an emulsion) is driven off through evaporation to form dry particles.
These particles can be processed further to form capsules or tablets. Product drying characteristics and powder requirements are considered while selecting the atomizer, temperature, optimal airflow pattern, and drying chamber configuration.
Adsorption on solid carriers
Formulations into free-flowing powders is through adsorption onto solid carriers. The procedure is simple you just mix the liquid formulation with a carrier material in a mixer. The powder that is formed can be filled directly into capsules or blended with other materials to form tablets.
One of the strengths of this approach is that it provides good uniformity in drug content. For example, a liquid SMEDDS formulation can be converted to a solid one by using maltodextrin as the carrier and the SMEDDS can be loaded on carriers at relatively high concentrations-up to 70% w/w.
The solid carrier materials that may be employed are microporous inorganic material, high-surface-area colloid material, cross-linked polymer, or nanoparticles. Silica, silicates, magnesium trisilicate, magnesium hydroxide, talc, crospovidone, aerosil 200 and cross-linked derivatives of sodium carboxymethyl cellulose or polymethyl methacrylate are some examples.
Cross-linked polymers are particularly beneficial as they serve to sustain drug dissolution over a period of time and minimize the likelihood of the drug re-precipitating. Nanoparticle carriers may consist of substances such as porous silicon dioxide, carbon nanotubes, carbon nano horns, fullerenes, charcoal, or even bamboo charcoal [48].
Melt granulation
A Less Complex Method of Producing Granules Melt granulation is a process that involves converting powders into granules by the incorporation of a binder that softens or melts upon gentle heat. The beauty of this process lies in the fact that it bypasses the traditional steps of incorporating liquid and drying-it is all one step. It is therefore quicker, cleaner, and an ideal solution when you don't wish to use solvents.
The process's success relies on several contributing factors such as the mixer speed, for how long you mix the ingredients, the particle size of the binder, and how sticky or thick the biner becomes when it melts.
There are also different solid and semi-solid lipids that have the ability to function as binders in melt granulation. One commonly utilized group is Gelucire®, which is a mixture of glycerides and PEG-based fatty acid esters. It's particularly good because, in addition to aiding in the formation of granules, it enhances the rate at which the drug is absorbed, due to its self-microemulsifying (SME) character.
Other lipid-based excipients such as lecithin, partial glycerides, and polysorbates have also been employed in this process. In most cases, the molten formulation (comprising the drug, lipids, and surfactants) is adsorbed onto inert solid carriers such as silica or magnesium alumina metasilicate to form a stable, solid SMEDDS formulation.
Melt extrusion
Melt extrusion is a neat, solvent-free technique that’s often used in pharmaceuticals. It’s especially useful because it can handle a high amount of drug (up to 60%) while keeping the mixture evenly distributed. The basic idea behind extrusion is to take a material that can be softened and shape it by pushing it through a die, kind of like squeezing toothpaste from a tube. This process is done under carefully controlled conditions, temperature, pressure, and flow to ensure consistent results [50].
The opening size of the die (or extruder) will influence how big the resulting pellets or spheroids turn out. One of the common ways this is used is in a method called extrusion spheronization, which is popular for making evenly sized pellets in drug manufacturing. The process involves several steps and they are
Blending
The L-SEDDS (which contains oil, surfactant, and drug) is mixed with solid carriers like polymers (e. g., PEG, PVP) or adsorbents (e. g., silica, magnesium aluminometasilicate). These carriers help solidify the liquid without compromising its ability to form microemulsions.
Heating and extrusion
The mixture is passed through a hot-melt extruder. Inside, heat softens the mixture, and mechanical force pushes it through a shaped die.
Cooling and shaping
As the extrudate exits the machine, it’s cooled and cut into pellets, granules, or filled into capsules, depending on the intended final dosage form.
Final product
The resulting solid SMEDDS can disperse in the gastrointestinal fluids to form a microemulsion, improving drug solubility and absorption just like the original L-SEDDS [51].
Benefits of using melt extrusion for S-SMEDDS
Avoids solvents (a solvent-free process)
Suitable for heat-stable drugs
Scalable for industrial production
Provides better stability and handling compared to liquid forms
CONCLUSION
Self-Microemulsifying Drug Delivery Systems (SMEDDS) are increasingly being employed to enhance the bioavailability of poorly water-soluble drugs. Despite their promise, a significant gap remains between the demand for effective lipid-based drug delivery systems and the currently available SMEDDS formulations. Most existing SMEDDS are formulated as liquid-filled soft gelatin capsules, which can lead to handling challenges, higher production costs, and stability concerns. To address these issues, there has been a growing focus on developing solid SMEDDS. Converting liquid SMEDDS into solid dosage forms can reduce handling difficulties, lower manufacturing costs, and improve product stability. Furthermore, it is essential that these formulations are designed to be compatible with the physiological environment to ensure optimal drug release and absorption. Continued research and development in this area will facilitate the broader application of SMEDDS, making this advanced drug delivery method more viable especially for drugs with poor aqueous solubility.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
All authors have contributed equally
CONFLICT OF INTERESTS
Declared none
REFERENCES
Anand S, Gupta R, Prajapathi SK. Self-microemulsifying drug delivery system. Asian J Pharm Clin Res. 2016;9(2):33-8.
Kyat Anwar AU, Jadav KR. Self-microemulsifying drug delivery system. J Pharm Res. 2010;3(1):75-83.
Mandawgade SD, Sharma S, Pathak S, Patravale VB. Development of SMEDDS using natural lipophile: application to β-artemether delivery. Int J Pharm. 2008;362(1-2):179-83. doi: 10.1016/j.ijpharm.2008.06.021, PMID 18652886.
Kang BK, Lee JS, Chon SK, Jeong SY, Yuk SH, Khang G. Development of self-microemulsifying drug delivery systems (SMEDDS) for oral bioavailability enhancement of simvastatin in beagle dogs. Int J Pharm. 2004;274(1-2):65-73. doi: 10.1016/j.ijpharm.2003.12.028, PMID 15072783.
Rahman MA, Hussain A, Hussain MS, Mirza MA, Iqbal Z. Role of excipients in successful development of self-emulsifying/microemulsifying drug delivery system (SEDDS/SMEDDS). Drug Dev Ind Pharm. 2013;39(1):1-19. doi: 10.3109/03639045.2012.660949, PMID 22372916.
Maurya SD, Arya RK, Rajpal G, Dhakar RC. Self-microemulsifying drug delivery systems (SMEDDS): a review on physicochemical and biopharmaceutical aspects. J Drug Delivery Ther. 2017;7(3):55-65. doi: 10.22270/jddt.v7i3.1453.
Khan AW, Kotta S, Ansari SH, Sharma RK, Ali J. Potentials and challenges in self-nanoemulsifying drug delivery systems. Expert Opin Drug Deliv. 2012;9(10):1305-17. doi: 10.1517/17425247.2012.719870, PMID 22954323.
Laddha P, Suthar V, Butani S. Development and optimization of self-microemulsifying drug delivery of domperidone. Braz J Pharm Sci. 2014;50(1):91-100. doi: 10.1590/S1984-82502011000100009.
Eccleston GM. Emulsions and microemulsions. In: Swarbrick J, editor. Encyclopaedia of Pharmaceutical Technology. New York: Informa Healthcare; 2007. p. 1548-65.
Mahmoud EA, Bendas ER, Mohamed MI. Preparation and evaluation of self-nanoemulsifying tablets of carvedilol. AAPS PharmSciTech. 2009;10(1):183-92. doi: 10.1208/s12249-009-9192-7, PMID 19238556.
Gao P. Design and development of self-emulsifying lipid formulations for improving oral bioavailability of poorly water-soluble and lipophilic drugs. In: Williams RO, Watts AB, Miller DA, editors. Formulating poorly water-soluble drugs. New York: Springer New York; 2012. p. 243-66. doi: 10.1007/978-1-4614-1144-4_7.
Nazzal S, Khan MA. Controlled release of a self-emulsifying formulation from a tablet dosage form: stability assessment and optimization of some processing parameters. Int J Pharm. 2006;315(1-2):110-21. doi: 10.1016/j.ijpharm.2006.02.019, PMID 16563673.
Gao P, Morozowich W. Development of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs. Expert Opin Drug Deliv. 2006;3(1):97-110. doi: 10.1517/17425247.3.1.97, PMID 16370943.
Huang Y, Tian R, Hu W, Jia Y, Zhang J, Jiang H. A novel plug-controlled colon-specific pulsatile capsule with tablet of curcumin loaded SMEDDS. Carbohydr Polym. 2013;92(2):2218-23. doi: 10.1016/j.carbpol.2012.11.105, PMID 23399280.
Woo JS, Song YK, Hong JY, Lim SJ, Choi HK. Reduced food effect and enhanced bioavailability of a self-microemulsifying formulation of itraconazole in healthy volunteers. Eur J Pharm Sci. 2008;33(2):59-65. doi: 10.1016/j.ejps.2007.11.001.
Taha E, Ghorab D, Zaghloul AA. Bioavailability assessment of vitamin a self-nanoemulsified drug delivery systems in rats: a comparative study. Med Princ Pract. 2007;16(5):355-9. doi: 10.1159/000104808.
Sander C, Holm P. Porous magnesium aluminometasilicate tablets as carrier of a cyclosporine self-emulsifying formulation. AAPS PharmSciTech. 2009;10(4):1388-95. doi: 10.1208/s12249-009-9340-0, PMID 19936938.
Khin Chi MP, Gupta A, Gupta MK. Self-emulsifying drug delivery system: a review. Asian J Biochem Pharm Res. 2011;1(2):359-67.
Bajaj H, Bisht S, Yadav M, Singh V. Bioavailability enhancement: a review. Int J Pharm Bio Sci. 2011;2(2):209-16.
Shukla JB, Koli AR, Ranch KM, Parikh RK. Self-microemulsifying drug delivery system. Int J Pharm Sci. 2010;1(2):13-33.
Shinde VJ, Orpe AA, Munde PV, Nimbalkar HY. Self-microemulsifying drug delivery system (SMEDDS): a novel approach. Am J Pharm Tech Res. 2020;10(1):251-65.
Manyam N. Recent advances in self-emulsifying drug delivery system. Int J Rev Life Sci. 2011;1(4):206-14.
Ingle LM. New approaches for development and characterization of SMEDDS. Int J Pharm Pharm Sci Res. 2013;3(1):7-14.
Agrawal S, Giri TK, Tripathi DK, Ajaz A, Alexander A. A review on novel therapeutic strategies for the enhancement of solubility for hydrophobic drugs through lipid and surfactant-based self-microemulsifying drug delivery system: a novel approach. Am J Drug Discov Dev. 2012;2(4):143-83. doi: 10.3923/ajdd.2012.143.183.
Patel D, Sawant KK. Self-microemulsifying drug delivery system: formulation development and biopharmaceutical evaluation of lipophilic drugs. Curr Drug Deliv. 2009;6(4):419-24. doi: 10.2174/156720109789000519, PMID 19534704.
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231-48. doi: 10.1038/nrd2197, PMID 17330072.
Chen ZQ, Liu Y, Zhao JH, Wang L, Feng NP. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int J Nanomedicine. 2012;7:1115-25. doi: 10.2147/IJN.S28761, PMID 22403491.
Bowtle W. Materials process and manufacturing considerations for lipid-based hard capsule formats. In: Hauss DJ, editor. Oral lipid-based formulations enhancing the bioavailability of poorly water-soluble drugs. New York: Informa Healthcare; 2007;170:79-106.
Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today. 2008;13(13-14):606-12. doi: 10.1016/j.drudis.2008.04.006, PMID 18598917.
Caliph SM, Charman WN, Porter CJ. Effect of short medium and long chain fatty acid-based vehicles on the absolute oral bioavailability and intestinal lymphatic transport of halofantrine and assessment of mass balance in lymph cannulated and non-cannulated rats. J Pharm Sci. 2000;89(8):1073-84. doi: 10.1002/1520-6017(200008)89:8<1073::aid-jps12>3.0.co;2-v, PMID 10906731.
Trevaskis NL, Charman WN, Porter CJ. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev. 2008;60(6):702-16. doi: 10.1016/j.addr.2007.09.007, PMID 18155316.
Ku MS, Li W, Dulin W, Donahue F, Cade D, Benameur H. Performance qualification of a new hypromellose capsule: part I. Comparative evaluation of physical, mechanical and processability quality attributes of Vcaps Plus quali-V and gelatin capsules. Int J Pharm. 2010 Feb 15;386(1-2):30-41. doi: 10.1016/j.ijpharm.2009.10.050.
Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today. 2008;13(13-14):606-12. doi: 10.1016/j.drudis.2008.04.006, PMID 18598917.
Zhang P, Liu Y, Feng N, Xu J. Preparation and evaluation of self-microemulsifying drug delivery system of oridonin. Int J Pharm. 2008;355(1-2):269-76. doi: 10.1016/j.ijpharm.2007.12.026, PMID 18242895.
Dahan A, Hoffman A. Rationalizing the selection of oral lipid-based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water-soluble drugs. J Control Release. 2008;129(1):1-10. doi: 10.1016/j.jconrel.2008.03.021, PMID 18499294.
Patil P, Patil V, Paradkar A. Formulation of a self-emulsifying system for oral delivery of simvastatin: in vitro and in vivo evaluation. Acta Pharm. 2007;57(1):111-22. doi: 10.2478/v10007-007-0009-5, PMID 19839411.
Tang JL, Sun J, He ZG. Self-emulsifying drug delivery systems: strategy for improving oral delivery of poorly soluble drugs. Curr Drug Ther. 2007;2(1):85-93. doi: 10.2174/157488507779422400.
Wasylaschuk WR, Harmon PA, Wagner G, Harman AB, Templeton AC, Xu H. Evaluation of hydroperoxides in common pharmaceutical excipients. J Pharm Sci. 2007;96(1):106-16. doi: 10.1002/jps.20726, PMID 16917844.
Sato K. Crystallization behaviour of fats and lipids a review. Chem Eng Sci. 2001;56(7):2255-65. doi: 10.1016/S0009-2509(00)00458-9.
Kovacic B, Vrecer F, Planinsek O. Spherical crystallization of drugs. Acta Pharm. 2012;62(1):1-14. doi: 10.2478/v10007-012-0010-5, PMID 22472445.
Mu H, Holm R, Mullertz A. Lipid-based formulations for oral administration of poorly water-soluble drugs. Int J Pharm. 2013;453(1):215-24. doi: 10.1016/j.ijpharm.2013.03.054, PMID 23578826.
Christensen KL, Pedersen GP, Kristensen HG. Technical aspects of spray drying and applications for the pharmaceutical industry. Pharm Dev Technol. 2001;6(3):277-87. doi: 10.1081/PDT-100107289.
Janga KY, Jukanti R, Vemula SK, Sunkavalli S, Velpula A, Bandari S. Solid self-emulsifying drug delivery systems: formulation characterization and biopharmaceutical performance. Curr Drug Deliv. 2012;9(3):222-32.
Tang B, Cheng G, Gu JC, Xu CH. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today. 2008;13(13-14):606-12. doi: 10.1016/j.drudis.2008.04.006, PMID 18598917.
Yi T, Wan J, Xu H, Yang X. A new solid self-microemulsifying formulation prepared by spray drying to improve the oral bioavailability of poorly water-soluble drugs. J Pharm Sci. 2008;97(2):646-59.
Ito Y, Kusawake T, Ishida M, Tawa R, Shibata N, Takada K. Oral solid gentamicin preparation using emulsifier and adsorbent. J Control Release. 2005;105(1-2):23-31. doi: 10.1016/j.jconrel.2005.03.017, PMID 15908031.
Kallakunta VR, Bandari S, Jukanti R, Veerareddy PR. Oral self-emulsifying powder of lercanidipine hydrochloride: formulation and evaluation. Powder Technol. 2012 May;221:375-82. doi: 10.1016/j.powtec.2012.01.032.
Ito Y, Kusawake T, Ishida M, Tawa R, Shibata N, Takada K. Oral solid gentamicin preparation using emulsifier and adsorbent. J Control Release. 2005;105(1-2):23-31. doi: 10.1016/j.jconrel.2005.03.017, PMID 15908031.
Venkatesan N, Yoshimitsu J, Ito Y, Shibata N, Takada K. Liquid-filled nanoparticles as a drug delivery tool for protein therapeutics. Biomaterials. 2005;26(34):7154-63. doi: 10.1016/j.biomaterials.2005.05.012, PMID 15967493.
Verreck G, Brewster ME. Melt extrusion-based dosage forms, excipients and processing conditions for pharmaceutical formulations. Bol Tech Gattefosse. 2004;97:85-95.
Clarke AP, Booth SW. Formulation variables on pellets containing self-emulsifying systems. Pharm Technol Eur. 2005;17(6):29-33.