
Int J Curr Pharm Res, Vol 17, Issue 5, 31-41Review Article
EXPLORING MODERN TECHNIQUES FOR SOLUBILITY ENHANCEMENT IN DRUG FORMULATIONS
PRIYANKA V. BAGADE1*, NILESH S. KULKARNI1, PRAVIN D. CHAUDHARI2, SHASHIKANT N. DHOLE1, UJWALA S. DESAI2
1*Progressive Education Society’s Modern College of Pharmacy (For Ladies) Moshi, Pune, Maharashtra, India. 2Progressive Education Society’s Modern College of Pharmacy Nigdi Pune, Maharashtra, India
*Corresponding author: Priyanka V. Bagade; *Email: priyankabagade25@gmail.com
Received: 08 Jun 2025, Revised and Accepted: 30 Jul 2025
ABSTRACT
By addressing the critical gaps in current practices, the objective of the review is to highlight emerging trends and provides insights into the optimization of drug solubility for improved pharmacological outcomes. The literature search was done and rlevent articles were collected from various database like Springer, Science Direct, Taylor and Francis, Wiley and pubmed.
Poor solubility of active pharmaceutical ingredients (APIs) significantly hinders their bioavailability and therapeutic efficacy, presenting a critical challenge in drug formulation. This review provides a comprehensive exploration of traditional and advanced techniques for solubility enhancement. Established methods, including salt formation, particle size reduction, and pH adjustment, are compared with cutting-edge strategies such as nanotechnology, electrospun nanofibers, spray drying, and supercritical fluid technology. These innovative approaches leverage mechanisms like particle size reduction, amorphization, and nanostructure engineering to enhance dissolution rates, stability, and controlled release profiles. The article discusses the Biopharmaceutical Classification System (BCS) and its relevance in tailoring solubility enhancement strategies for poorly water-soluble compounds.
Keywords: Solubility enhancement, Drug bioavailability, Nanotechnology-based formulations, Pharmaceutical product development
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijcpr.2025v17i5.7044 Journal homepage: https://innovareacademics.in/journals/index.php/ijcpr
INTRODUCTION
Solubility plays a critical role in the pharmacological efficacy of active pharmaceutical ingredients (APIs), as it directly impacts the absorption and bioavailability of drugs [1]. Pharmaceutical substances that are poorly soluble in water phase significant challenges, including inconsistent absorption and variable therapeutic outcomes, particularly when administered orally [2]. This issue is increasingly prevalent with the growing complexity of modern drug candidates, which often exhibit higher lipophilicity, larger molecular sizes, and reduced aqueous solubility. As a result, the development of poorly water-soluble drugs has become a major hurdle in pharmaceutical research and formulation [3]. It is estimated that approximately 40% of the top 200 oral medications marketed in the United States, 33% of drugs in the US pharmacopoeia, 75% of compounds in development, and 90% of newly discovered chemical entities suffer from poor water solubility [1]. This widespread issue not only leads to limited bioavailability but also necessitates higher drug doses to achieve therapeutic effects, thus increasing the risk of adverse side effects and decreasing patient compliance. The Biopharmaceutical Classification System (BCS) classifies drugs based on their solubility and permeability characteristics, and many poorly soluble drugs fall into BCS Classes II and IV, which further complicates their development and effective therapeutic application [3].
To address these challenges, a variety of solubility enhancement techniques have been developed. Traditional methods, such as salt formation, particle size reduction, and the use of surfactants, have been employed for decades with varying degrees of success. However, these techniques often fail to fully address the complexities of modern poorly soluble compounds. Recent innovations in pharmaceutical formulation have introduced more advanced strategies, including nanocrystals, amorphous solid dispersions, cyclodextrin complexes, and nanotechnology-based systems [4]. Other promising techniques such as electrospun nanofibers, spray drying, supercritical fluid technology, and self-microemulsifying drug delivery systems (SMEDDS) provide new possibilities for improving solubility, dissolution, and bioavailability. These advanced methods not only enhance solubility but also offer controlled release, improved stability, and more predictable therapeutic outcomes [5]. This review explores the various solubility enhancement techniques, focusing on both traditional and innovative approaches. It provides a systematic classification of these methods, their respective advantages and limitations, and their applications in overcoming the solubility challenges encountered in pharmaceutical development. By examining these strategies, this review aims to provide a comprehensive understanding of solubility enhancement and its crucial role in improving drug formulation and therapeutic efficacy.
Classification of solubility enhancement techniques
The techniques for solubility improvement can be grouped into four main categories: chemical modifications, physical modifications, powder granulation methods, and miscellaneous approaches [6-9].
Physical methods
Particle size reduction is a foundational strategy for enhancing the solubility of poorly water-soluble drugs. By decreasing the size of drug particles, the surface area increases significantly, promoting faster dissolution rates [10]. While modern advancements have introduced nanotechnology-based methods, conventional mechanical size reduction techniques remain widely used due to their simplicity and effectiveness. Below is an exploration of these conventional methods:
Mechanical micronization
Micronization involves reducing drug particles to sizes in the range of 2–5 µm, which enhances dissolution by increasing the surface area-to-volume ratio [11]. Common techniques include:
Jet milling
It is a widely used technique for reducing particle size to enhance drug solubility and dissolution. It uses high-velocity air streams to accelerate drug particles, causing collisions that lead to size reduction through impact and attrition. This process typically reduces particle sizes from 20–100 µm to less than 10 µm, making it suitable for heat-sensitive drugs and materials prone to melting [12]. The integration of classifiers ensures precise particle size control, while longer milling durations yield finer particles. Fluid energy milling has been effectively applied to improve the dissolution rates of drugs like ibuprofen [13], salbutamol sulfate [14], and fenoterolhydrobromide [15]. Co-milling with hydrophilic excipients has shown further improvements in dissolution, as seen with fenofibrate and EMD (Include full form) 57033 [16]. The technique also supports surface modifications, such as coating milled particles with nanosilica to enhance flow properties and reduce agglomeration. Although newer methods, such as wet milling, produce finer particles at the nanoscale, fluid energy milling remains a benchmark for evaluating and developing advanced milling strategies [17]. Studies demonstrate that controlling the crystal size and morphology of starting materials can significantly influence milling outcomes, making fluid energy milling comparable to wet milling for specific applications [18]. Additionally, it serves as a preparatory step for nanoparticle production by integrating with processes like high-pressure homogenization.
Ball milling
Employs grinding media such as ceramic or steel balls to break down particles. This method is also capable of producing amorphous forms of drugs when combined with polymers, which can further enhance solubility. Research on drugs like danazol has demonstrated increased bioavailability using ball milling [19]. Solid dispersions of etodolac (ETD) prepared using ball milling demonstrated significantly enhanced solubility and faster dissolution compared to the unprocessed drug. Amorphous carriers (e. g., HPMC, PVP) outperformed crystalline carriers (e. g., urea, mannitol) in improving these properties [20].
High-pressure homogenization (HPH)
HPH is a top-down approach that reduces particle sizes to the nanoscale by forcing a drug suspension through a high-pressure valve. The sudden pressure drop induces cavitation, turbulence, and collision, effectively breaking down particles. This method is widely used for both oral and parenteral formulations. Examples include improved dissolution of prednisolone and carbamazepine using HPH. Recently published data demonstrated that combining antisolvent crystallization with high-pressure homogenization significantly improves the solubility and dissolution of curcumin, with PVP-K30 as an effective stabilizer, enhancing solubility up to 25-fold by reducing particle size and promoting amorphous formation. Another advantages of HPH include its ability to maintain drug stability without introducing contaminants, making it suitable for sterile formulations [21].
Cryogenic grinding
Cryogenic grinding involves freezing drugs with cryogens like liquid nitrogen before mechanical grinding. This prevents heat-induced degradation and achieves finer particles. It is particularly beneficial for thermosensitive drugs, enabling solubility enhancement without altering chemical properties. Recent research highlights the successful application of cryogenic grinding to convert crystalline drugs into their amorphous forms, enhancing dissolution rates and stability [22]. For instance, amorphization was observed in various polymorphs of indomethacin, including γ-indomethacin, where cryomilling improved dissolution rates. Studies also examined the physical stability of cryomilled amorphous indomethacin, tracking its re-crystallization during storage [23]. Similarly, cryogenic grinding effectively transformed crystalline glibenclamide into its amorphous form without inducing chemical degradation, a process linked to its amide-imidic acid tautomerism [24].
Crystal engineering
It focuses on manipulating the crystalline structure of drug particles to improve solubility, dissolution rate, and bioavailability. It involves altering the solid-state properties of drugs through techniques such as polymorphism, co-crystallization, and amorphization [25].
Polymorphism: Polymorphs are different crystalline forms of the same drug molecule. Metastable polymorphs typically have lower lattice energy, leading to better solubility compared to stable forms.
Co-crystallization: This involves forming a crystalline complex between the active pharmaceutical ingredient (API) and a co-former, which can enhance solubility and stability without altering the drug's chemical structure. Drug–drug cocrystallization of carvedilol with hydrochlorothiazide improved solubility by 7.3 times and dissolution by 2.7 times, with stability over 90 days, offering a promising approach to enhance bioavailability [26].
Amorphization: Amorphization is an effective strategy to enhance the solubility and dissolution rate of poorly water-soluble drugs by disrupting their crystalline structure. For instance, cryomilling γ-indomethacin transforms it into an amorphous form, resulting in a significant increase in solubility and dissolution, which improves with milling duration [27, 28]. Crystal engineering technologies, including nanocrystals and engineered co-crystals, are valuable for optimizing drug solubility while maintaining stability. These approaches are highly versatile and can be tailored to address specific formulation challenges.
Chemical methods
Chemical methods such as salt formation, co-crystallization, pH adjustment, and complexation with agents like cyclodextrins play a significant role in modifying the physicochemical properties of drugs [29].
pH modification
An effective method to enhance the solubility of pH-dependent, poorly soluble drugs is through pH adjustment. This involves incorporating pH-modifying agents into formulations or tablets to create a microenvironment with an optimal pH, thereby improving drug solubility and release [30]. For instance, weakly basic drugs with low solubility can benefit from the use of acidifiers, while weakly acidic drugs can be solubilized using alkaline agents. However, this approach has limitations, particularly when high drug load formulations require significant quantities of pH modifiers [31]. This can compromise stability, especially if hygroscopic agents like sodium hydroxide are used. Thus, the choice of pH modifier is critical to achieving a balance between enhanced solubility and maintaining formulation stability. In recently published work, alkaline agents such as meglumine, sodium carbonate, and Neusilin S2 were selected to evaluate their ability to create a favorable microenvironmental pH and enhance drug solubility and dissolution rates [32]. The study examined how pH modifiers enhance the solubility, dissolution, and stability of telmisartan (TEL) solid dispersions made via hot-melt extrusion. Meglumine (MEG) and sodium carbonate (SC) improved solubility by 9-fold (1.15 mg/ml) and 7-fold (0.96 mg/ml), respectively. Another study demonstrated that combining posaconazole (POS) with citric acid (CA) enhances solubility and dissolution by promoting amorphization and creating an acidic environment [33]. The POS-CA system showed superior performance in solubility, dissolution, and stability compared to the POS-VA64 formulation. This highlights the effectiveness of pH modification for improving drug properties.
Salt formation
The solubility of salts is governed by the pH–solubility profile [34], where basic drugs form salts below their pHmax, and acidic drugs above their pHmax. For example, the solubility of a basic drug salt can be expressed as

Where Ka is the acid dissociation constant, [BH+]s is the saturation concentration of salt.
Key factors influencing solubility include pKa, solubility product (Ksp), and counterion choice, as stronger acids (e.g., HCl or methanesulfonic acid) tend to produce salts with higher solubility [35]. However, challenges such as the common-ion effect—where excess counterions reduce solubility—and salt conversion to free acid or base forms under gastrointestinal pH conditions can impact dissolution and bioavailability. Non-HCl salts like mesylates are increasingly favored for overcoming common-ion effects[36]. Effective salt screening based on pH–pH-solubility principles minimizes trial-and-error experimentation, ensuring the selection of optimal salts for both liquid and solid dosage forms, thereby maximizing solubility, dissolution rate, and therapeutic efficacy.
Recently published work explores how salt formation can improve the solubility and dissolution properties of poorly soluble drugs, using benexate salts with artificial sweeteners such as saccharinate and cyclamate [37]. These salts showed significant enhancements in solubility (up to fivefold) and faster dissolution rates compared to the original drug.
Complexation
It involves the formation of stable complexes between a drug molecule (substrate) and a ligand, which can occur through various interactions such as hydrogen bonding, hydrophobic forces, van der Waals interactions, and electrostatic interactions [38]. Cyclodextrins (CDs) are among the most widely used complexing agents due to their ability to encapsulate hydrophobic drug molecules within their hydrophobic core while exposing hydrophilic surfaces to the aqueous environment [39]. This structural property significantly improves the solubility of hydrophobic drugs. For instance, β-CD complexes of norfloxacin [40] and curcumin have demonstrated remarkable solubility enhancements [41]. The conversion of crystalline drugs into amorphous forms during complexation is another critical factor that enhances dissolution rates. Additionally, techniques such as kneading, coprecipitation, solvent evaporation, and freeze-drying are commonly employed to prepare drug complexes, ensuring uniformity and efficiency in drug formulation [42]. Curcumin-CD complexes have shown a 20-fold increase in solubility and a six-fold improvement in bioavailability [41], making them more effective for oral administration and anticancer therapies. Similarly, inclusion complexes of taxifolin-γ-CD and ibuprofen-CD not only improved solubility but also exhibited faster drug release profiles due to reduced particle sizes and enhanced hydrophilicity [43]. Moreover, another study focused on improving the solubility of mefenamic acid (MA), a poorly water-soluble drug, by forming inclusion complexes with β-cyclodextrin (β-CD). The 2:1 MA: β-CD complex showed significantly enhanced solubility, with the solvent co-evaporation (CE) method providing the best dissolution improvement [44].
Miscellaneous methods
Supercritical fluid technology, micellar solubilization, and hydrotropy are innovative strategies for improving the solubility of poorly water-soluble drugs, a common challenge in pharmaceutical development. Supercritical fluids, such as carbon dioxide, operate at conditions above their critical temperature and pressure, offering unique properties that allow for the dissolution and precise recrystallization of drug particles into nanometer or submicron sizes, which enhances their solubility and bioavailability [45-47].
Supercritical fluid technology
Supercritical fluid (SCF) technology is an advanced and environmentally sustainable approach to enhancing the solubility of poorly water-soluble drugs, a critical challenge in pharmaceutical formulation. SCFs, particularly supercritical carbon dioxide (SC-CO2), operate at conditions above their critical temperature and pressure, giving them properties that are intermediate between those of liquids and gases [48, 49]. SCF technology, particularly the Supercritical Anti-Solvent (SAS) process, significantly enhances the solubility of poorly water-soluble drugs like betamethasone by reducing particle size and converting drugs to an amorphous state. This results in a 2.8–3.5fold increase in solubility and rapid drug release within 60 min. SCF methods maintain drug integrity, improve biocompatibility, and offer a scalable, eco-friendly solution for advanced drug delivery systems [50, 51]. Additionally, SCF-based methods often result in the amorphous form of the drug, which dissolves more rapidly than its crystalline counterpart due to its higher energy state and greater solubility [52]. Moreover, SCF technology is instrumental in enhancing the performance of drug-polymer systems. SCFs can penetrate polymer matrices and facilitate uniform drug dispersion, improving the carrier's encapsulation efficiency. This, coupled with the reduced particle size, enhances the drug's wettability and dissolution characteristics [53]. For example, drugs such as curcumin and 5-fluorouracil have shown significant solubility and bioavailability improvement when processed using SCF techniques [45].
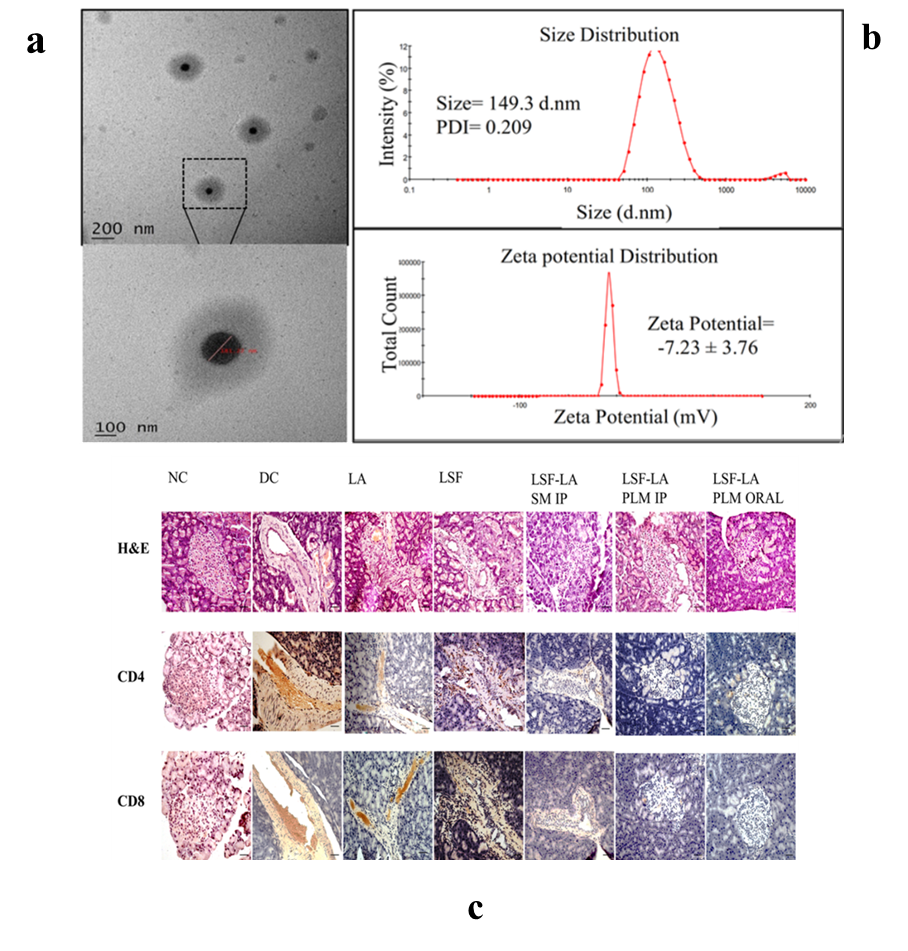
Fig. 1: (a) High-resolution TEM images illustrating the particle size morphology of LSF−LA PLM; (b) Particle size distribution and zeta potential analysis of LSF−LA PLM; (c) Histological and immunohistochemical analysis of pancreatic sections after 21 d of treatment: H and E staining and expression of CD4+and CD8+T-cells [56]
Micellar solubilization
Micellar solubilization, also known as solubilization by surfactants, involves the use of amphiphilic molecules containing both polar and non-polar regions to enhance the solubility of poorly water-soluble drugs. Studies demonstrated significant solubility enhancement for various poorly soluble drugs, including several BCS class II antidiabetic drugs like glyburide, glimepiride, and pioglitazone [54]. This approach has been widely reviewed and validated for its efficiency in improving drug solubility and bioavailability. Moreover, recently published study improved the solubility and stability of caffeic acid (CA), a hydrophobic polyphenol, by incorporating it into micelles [55]. The micelles, sized 11.70–17.70 nm, enhanced solubility primarily through micellar solubilization, where hydrophobic CA molecules were encapsulated in the micelle's hydrophobic core, shielding them from the aqueous environment. The recent study developed a polymeric micellar formulation to improve the oral delivery of Lisofylline (LSF), an anti-inflammatory drug with potential for type 1 diabetes (T1D) treatment [56]. Encapsulation of LSF’s linoleic acid prodrug in biodegradable micelles (~149.3 nm) (fig. 1). Histopathological analysis showed preserved pancreatic β-cells and reduced immune cell infiltration (CD4+and CD8+T-cells) (fig. 2c).
Hydrotropy
Hydrotropy as a sustainable and eco-friendly method for enhancing the solubility of poorly water-soluble pharmaceutical active ingredients (PAIs). Hydrotropes, which are amphiphilic molecules, enhance solubility through weak interactions like hydrogen bonding and π–π stacking without forming micelles, distinguishing them from surfactants.
Hydrotropy supports sustainability goals by minimizing hazardous waste, energy consumption, and environmental impact, making it a promising approach for pharmaceutical and analytical applications. Seishi Shimizu and Nobuyuki Matubayasi developed a theoretical model grounded in statistical thermodynamics to explain the cooperative behavior observed in hydrotropic solubilization [57]. They demonstrated that solubility exhibits a sigmoidal pattern, characterized by a sharp increase after reaching the minimum hydrotrope concentration (MHC) and eventually stabilizing at higher concentrations. Among the studied hydrotropes, GVL was particularly effective in high-concentration regions, demonstrating a balanced interaction between solute-hydrotrope and hydrotrope-hydrotrope systems [58]. These findings underline the potential of biobased hydrotropes as sustainable, eco-friendly alternatives for enhancing the solubility of hydrophobic compounds in water, offering practical applications in pharmaceuticals and other industries. Another study explores the use of amino acids as hydrotropes to enhance the solubility of poorly soluble drugs, carbamazepine (CBZ) and indomethacin (IND). Neutral hydrophobic amino acids improved IND solubility through non-ionic interactions, while both neutral and charged amino acids enhanced CBZ solubility via hydrophobic interactions and hydrogen bonding. A combination of multiple amino acids led to up to a 7-fold increase in IND solubility. The findings highlight the potential of amino acids to improve the aqueous solubility of poorly soluble drugs, which is important for pharmaceutical development.
Innovative solubility enhancement techniques
Innovative solubility techniques are summarized below
Nanotechnology approaches
Polymeric nanoparticles
Enhancing the solubility of poorly water-soluble drugs is crucial for improving their bioavailability, as over 40% of new drug candidates face this limitation. For polymeric drug delivery systems, hydrophilic polymers, monomers, and co-polymers present the most promising solutions [59]. These hydrophilic materials, such as β-cyclodextrins (β-CD), chitosan, chondroitin sulfate, polyethylene glycols (PEGs), poloxamer, sodium alginate, 2-hydroxyethyl starch, and polyvinyl pyrrolidone (PVP), can achieve up to 90% hydration, significantly outperforming hydrophobic polymers, which offer only 5–10% hydration [6]. The study explores polymeric nanoparticles prepared via sonoprecipitation to address solubility challenges, using curcumin (CUR) as a model drug [60]. Sonoprecipitation combines ultrasonic energy with precipitation to reduce particle size and modify molecular interactions, thereby improving dissolution rates. Smaller nanoparticles, achieved using hydrophilic hydroxypropyl methylcellulose (HPMC), significantly enhance solubility [61]. Moreover, the method converts crystalline CUR to an amorphous state, promoting higher dissolution rates. Intermolecular hydrogen bonding between CUR and HPMC 6 stabilizes the amorphous phase and facilitates drug release. Optimizing ultrasonication conditions, such as duration and power, and the drug-to-polymer ratio, was critical for achieving desirable outcomes. Longer ultrasonication times and higher power produced finer particles and stronger drug-polymer interactions. Increasing the polymer content further reduced particle size and improved steric stabilization, enhancing the dissolution profile [62]. The superior hydration properties of hydrophilic polymers like HPMC contributed significantly to these results. Overall, the study highlights polymeric nanoparticles and the sonoprecipitation method as effective strategies for enhancing solubility and bioavailability, providing a scalable and cost-effective solution for poorly water-soluble drugs. Similarly, in another work demonstrated that Cellulose acetate-based polymeric nanoparticles significantly enhanced the solubility and dissolution of poorly soluble antiviral drugs ritonavir and efavirenz. Produced via rapid precipitation, these 150–200 nm nanoparticles achieved 88–96% drug loading efficiency and stabilized the drugs in an amorphous state, as confirmed by XRD and DSC. Dissolution studies showed a 10–20-fold increase in solubility, with CMCABdelivering the highest release (up to 40%). The combined effects of particle size reduction and recrystallization inhibition by the polymers underscore their potential for improving drug bioavailability [63].
Solid lipid nanoparticles (SLNs)
Composed of biocompatible and physiological lipids stabilized by surfactants, SLNs improve drug solubility by encapsulating hydrophobic drugs in their lipid matrix. Solid lipid nanoparticles (SLNs) play a significant role in enhancing the solubility and bioavailability of poorly water-soluble drugs [64]. These lipid-based carriers improve drug absorption by facilitating transport through specialized pathways in the gastrointestinal tract. SLNs are absorbed via mechanisms like transcellular and paracellular pathways, with transcellular uptake involving energy-dependent endocytosis processes such as clathrin-mediated, caveolin-mediated, and macropinocytosis [65]. In addition, SLNs can target M cells in Peyer’s patches for efficient transcytosis, bypassing enzymatic degradation in the gut. Once absorbed, lipid-based SLNs can promote the formation of chylomicrons by enterocytes, facilitating lymphatic transport and avoiding hepatic first-pass metabolism [66]. Similarly, Nimodipine (NMD), an antihypertensive drug with low oral bioavailability, was encapsulated in solid lipid nanoparticles (NMD-SLNs) to enhance absorption and target the intestinal lymphatic system [67]. The nanoparticles were formulated using palmitic acid, poloxamer 188, and soya lecithin. Optimized NMD-SLNs exhibited a particle size of 116 nm, high drug entrapment (93.66%), and sustained release (87.52% in 10 h). In vivo studies in rats showed a 2.08-fold increase in bioavailability compared to NMD solution. Stability tests revealed no significant changes after 3 mo, indicating the potential of NMD-SLNs for improved oral delivery. Similarly, in another research, Nitrendipine, an antihypertensive drug with poor oral bioavailability (10-20%) due to first-pass metabolism, was formulated into solid lipid nanoparticles (SLNs) to enhance absorption [68]. Using tripalmitin, glyceryl monostearate, and cetyl palmitate as lipids and poloxamer 188 as a surfactant, SLNs were prepared via hot homogenization and ultrasonication. The nanoparticles were characterized for particle size, zeta potential, and entrapment efficiency, with in vitro release studies conducted in phosphate buffer (pH 6.8). Pharmacokinetic studies in Wistar rats showed a 3 to 4 fold increase in bioavailability compared to a nitrendipine suspension, highlighting SLNs' potential to enhance bioavailability by reducing first-pass metabolism.
Nanoemulsions
Nanoemulsions are specialized colloidal systems designed to improve the solubility and bioavailability of drugs with poor water solubility. Comprising nanosized droplets stabilized by surfactants and co-surfactants, they offer enhanced surface area for drug dissolution, thereby facilitating better absorption [69]. Preparation methods such as ultrasonication and high-pressure homogenization create uniform and stable droplets, while low-energy approaches provide simpler alternatives. These systems are especially beneficial for delivering hydrophobic drugs, as they protect the active ingredients from enzymatic breakdown and improve systemic uptake [70]. Research on nanoemulsions, such as those developed for artemether, highlights their ability to significantly enhance drug release and bioavailability compared to traditional formulations [71]. However, nanoemulsions must address challenges like thermodynamic instability, which can cause droplet coalescence or phase separation. Stability can be improved by using high-viscosity oils or polymers, such as poly(δ-decalactone), which minimize issues like Ostwald ripening and oxidation [72]. Their versatility makes nanoemulsions suitable for various delivery methods, including oral [73], parenteral [74], and ophthalmic applications [75]. These formulations enhance drug solubility and absorption, provide controlled release, and improve therapeutic efficacy, making them a promising advancement in pharmaceutical technology. Nanoemulsions formulated with clove oil and isopropyl myristate as the oil phase, combined with Tween 80 and isopropyl alcohol as surfactant and co-surfactant, demonstrated promising properties for enhancing drug delivery [76]. Diflunisal and niflumic acid nanoemulsions exhibited hydrodynamic droplet sizes between 8 and 22 nm with a polydispersity index below 0.5, indicating a uniform distribution. Although low zeta potentials suggested metastable formulations, accelerated stability tests confirmed their stability under various conditions. Another work demonstrated nanoemulsions (NE) for oral delivery of low molecular weight heparin (LMWH). Non-digestible NE with polyglycerol-, PEGylated, or zwitterionic surfactants were tested, incorporating LMWH via hydrophobic ion pairing (HIP) [77]. The formulations showed droplet sizes below 150 nm, with polyglycerol-surfactant NE being less toxic and hemolytic than PEGylated or zwitterionic NE. Non-digestible NE provided greater stability and improved LMWH bioavailability up to 2.6% in vivo, demonstrating their superiority over biodegradable formulations for oral LMWH delivery. These nanoemulsions show potential for improving the bioavailability and therapeutic efficacy of poorly water-soluble drugs across multiple administration routes, including oral, transdermal, and localized applications.
Liposomes
Liposomes are vesicles composed of self-assembling lipid bilayers made of phospholipids and cholesterol. They are classified into large unilamellar vesicles (LUV), small unilamellar vesicles (SUV), multilamellar vesicles (MLV), and multivesicular vesicles (MVV). LUV, SUV, and MLV can be used for various delivery routes, including oral, while MVV are suited for parenteral delivery. Liposomes can encapsulate hydrophilic drugs in their inner aqueous phase and hydrophobic drugs within their lipid bilayer, making them versatile drug delivery systems. Their biphasic nature and customizable structure provide significant potential for improving drug solubility and delivery [78]. Encapsulation efficiency for lipophilic drugs can reach up to 100% when optimized with lipophilic derivatives and appropriate bilayer composition [79]. However, drug incorporation is influenced not only by the physicochemical properties of the drug but also by the liposomal bilayer composition and the preparation method, making them a versatile tool for enhancing drug solubility and bioavailability. For poorly soluble drugs, liposomes are particularly beneficial as they enhance solubility, protect the drug from premature degradation, and improve systemic absorption [80]. The drug release mechanism from liposomes primarily depends on two pathways: diffusion through the aqueous phase and collisions between liposomes. In the diffusion process, drugs are released directly into the surrounding aqueous medium, migrating from donor to acceptor liposomes. This mechanism is influenced by factors such as the solubility of the drug, the stability of the liposome, and the chemical properties of the lipid bilayer. In contrast, in the collision-based transfer, physical interactions between liposomes facilitate the direct migration of drug molecules from one liposome to another. These processes often exhibit first-order kinetics, characterized by exponential release behavior, particularly when liposome loading is low and drug interactions within the bilayer are minimal [81]. Research has shown that liposomes combined with excipients such as cyclodextrins and meglumine significantly increase drug encapsulation and solubility. For instance, sulfamerazine and indomethacin, poorly soluble drugs, showed enhanced incorporation efficiency and stability when included in liposomal formulations with β-cyclodextrins or hydroxypropyl-β-cyclodextrins. These systems improved drug solubilization up to 18-43 times compared to conventional liposomes, highlighting the effectiveness of these modifications in addressing solubility challenges. Moreover, the controlled release properties of liposomes make them effective in maintaining drug stability and prolonging therapeutic effects. The release mechanism is influenced by drug-lipid interactions, preparation methods, and the incorporation of excipients like polyethylene glycol (PEG), which can modulate release kinetics and improve circulation time. Advanced techniques such as thin-film hydration and dehydration-rehydration have also enhanced encapsulation efficiency and delivery precision [82]. Berberine hydrochloride (BBR), a plant-derived alkaloid, faces solubility issues due to its low water solubility, which limits its bioavailability and therapeutic effectiveness. To address this, liposomal delivery systems have been developed [83], but traditional liposomes suffer from poor stability and rapid clearance. This study presents Fiber Interlaced Liposomes™ (FIL), which use plant-based fibers to enhance liposome stability, solubility, and protect against degradation. The FIL-BBR formulation demonstrated sustained drug release and improved stability in simulated gastric and intestinal fluids. In vivo studies in rats showed a 3.37 fold increase in oral bioavailability, with higher AUC and Cmax compared to unformulated BBR. These results highlight the potential of FIL-based liposomes for improving BBR’s solubility, stability, and bioavailability.
Electrospun technology
Electrospun nanofibers have emerged as an innovative solution for enhancing the solubility and bioavailability of poorly water-soluble drugs [84]. Produced through electrospinning, these nanofibers offer a high surface area-to-volume ratio, porosity, and tunable composition, making them ideal for improving drug dissolution [85]. By dispersing drugs in an amorphous or nanocrystalline state within the polymer matrix, nanofibers eliminate the crystalline structure that limits solubility, leading to faster dissolution rates. Incorporating solubility-enhancing excipients like cyclodextrins or surfactants further boosts solubility by forming drug-inclusion complexes and stabilizing the amorphous form. Additionally, polymers such as polyvinylpyrrolidone (PVP) and polyethylene oxide (PEO) enhance dissolution and prevent recrystallization. The release profile can be tailored, enabling sustained or immediate drug release, with advanced designs like core-shell nanofibers offering controlled delivery [84]. This versatility and efficiency make electrospun nanofibers a promising platform for addressing solubility challenges and achieving improved therapeutic outcomes. Recently published work demonstrated preparation of optimized fast-dissolving delivery systems (FDDS) composed of nanofiber mats fabricated from PVP and containing PCPZ for oromucosal application [86]. The optimized formulation achieved 88.02% drug entrapment, rapid disintegration (<1 second), and drug release of 91.49% within 2 min, outperforming commercial Stemetil MD® tablets. The nanofibers displayed high permeability across the oromucosal membrane (31.28 μg) and reduced crystallinity, enhancing solubility and bioavailability. With suitable physio-mechanical properties, biocompatibility, and early onset of action, the nanofiber formulation presents a promising oromucosal delivery system for prochlorperazine. Similarly, to overcome the issue of Curcumin's hydrophobicity and poor stability, fast-dissolving drug delivery systems (FDDDS) were developed using curcumin-loaded JFP/Pullulan nanofibers via electrospinning [87]. Ultrasonication produced the most stable emulsion with uniform droplet size (365.8 nm) and PDI of 0.174. The 95:5 formulation exhibited high encapsulation efficiency (83.98%) and a nanofiber diameter of 104.2 nm. These nanofibers showed enhanced antioxidant activity, rapid disintegration in artificial saliva, and improved curcumin solubility and stability, offering a promising drug delivery platform. Similar to curcumin, Diosmetin (DT), known for its anticancer, antibacterial, antioxidant, and anti-inflammatory properties, faces challenges in therapeutic use due to its poor water solubility [88]. To address this, carboxymethyl cellulose/polyvinyl alcohol (CMC/PVA) nanofibers loaded with DT were fabricated via electrospinning. The resulting nanofibers (P11C2DT) exhibited a uniform size of 151 nm, smooth morphology, and 82.8% drug loading efficiency. Characterization through SEM, FT-IR, XRD, TGA, and DSCconfirmed the nanofibers' structural and thermal properties. Notably, the nanofibrous membrane dissolved 85% of DT in the release medium within 5 h, a significant improvement compared to the undissolved powdered DT, highlighting its potential to enhance DT's solubility and therapeutic applicability. Another study explored the use of electrospun nanofibers made from polyvinyl pyrrolidone (PVP) to enhance the dissolution rate of piroxicam, a poorly water-soluble drug [89]. Piroxicam was incorporated into PVP nanofibers at a 1:4 drug-to-polymer ratio, resulting in fibers with a uniform size (600 nm) (fig. 2). Dissolution tests showed that the nanofibers released 91.32% of the drug in 30 min, significantly higher than the 63.35% released by the pure drug. This improvement is due to the increased surface area and the amorphous nature of the drug in the nanofiber matrix, highlighting electrospun nanofibers as an effective method for enhancing drug solubility and bioavailability.

Fig. 2: SEM images of drug-loaded nanofibers: (a, b) Nanofibers without drug loading; (c) PVP nanofibers; (d, e, f) PVP nanofibers loaded with piroxicam at increasing magnifications
Spherical crystallization/spherical agglomeration
Spherical crystallization has gained significant attention in the pharmaceutical, chemical, and food industries due to its benefits in enhancing the properties of powders. This technique combines crystallization, grinding, and granulation, simplifying the drug formulation process, reducing production time, and lowering costs [90]. Spherical drug particles offer advantages such as improved compressibility, anti-caking properties, mechanical strength, and enhanced dissolution rates. Additionally, spherical agglomerates of multi-component drugs contribute to better morphology and uniform distribution, improving patient compliance and bioavailability. Among the various spherical crystallization methods, spherical agglomeration (SA) and crystal co-agglomeration (CCA) are the most widely used. SA, which relies on a mixed solvent system, involves crystallization, phase separation, wetting, and agglomeration steps, influenced by factors like solvent system, temperature, and agitation [91]. Compared to traditional granulation methods, SA and CCA offer simpler, more cost-effective processes for improving the solubility, flowability, and compressibility of poorly soluble drugs. Recently published study investigates the use of spherical crystallization to enhance the solubility, micromeritics, and compactability of oxcarbazepine (OXZ), an anticonvulsant drug [92]. Spherical agglomerates were prepared using different polymers (PEG 6000 and PVP K30) and various process parameters, utilizing water, dichloromethane, and chloroform as solvents. The agglomerates showed significant improvements in solubility, dissolution rate, and micromeritics compared to pure OXZ. Characterization through techniques like FTIR, DSC, SEM, and XRPD revealed that the agglomerates were more amorphous, with reduced crystallinity, leading to enhanced bioavailability. The results suggest that spherical agglomeration can serve as an effective alternative to traditional granulation for preparing BCS class II drugs with improved physicochemical and bio-pharmaceutical properties [92]. Another research demonstrated development of polymeric spherical agglomerates of Bosentan monohydrate using the crystallo-co-agglomeration (CCA) technique, enhancing solubility, micrometric properties, and dissolution rate [93]. Optimization via Box–Behnken design identified talc and PEG6000 concentrations, along with rotation speed, as critical factors. Characterization confirmed improved sphericity, flowability, and conversion from crystalline to amorphous form, with no drug-excipient interactions. Fast dispersible tablets (FDTs) formulated from the agglomerates showed significantly enhanced drug release (94.14% in 20 min) and maintained stability over three months. This approach demonstrates potential for improving the performance of poorly soluble drugs.
Liquisolid technique
Liquisolid technology has emerged as a promising approach to improve the solubility, dissolution, and bioavailability of poorly water-soluble drugs, particularly those in BCS Classes II and IV. This technique converts liquid medications, such as drug solutions or suspensions, into free-flowing and compressible powders [94]. It utilizes non-volatile liquid vehicles like polyethylene glycol (PEG) and propylene glycol to dissolve or disperse the drug, which is then adsorbed onto porous carriers such as microcrystalline cellulose (MCC) or starch and coated with materials like silica to enhance flowability. The molecular dispersion of drugs within this system significantly increases surface area, wettability, and dissolution rate, enabling rapid drug release in dissolution media [95]. Furthermore, the liquisolid technique can protect light-sensitive drugs, improve flow properties, and facilitate the development of both immediate-and sustained-release formulations, offering a versatile solution for addressing solubility challenges.
Recent research highlights the effectiveness of liquisolid technology in enhancing the performance of hydrophobic drugs. For instance, nimodipine, a poorly soluble antihypertensive drug, exhibited significantly improved dissolution and bioavailability when formulated into liquisolid compacts with non-volatile solvents and advanced coating agents like Neusilin [96]. Similarly, curcumin, a hydrophobic compound with poor bioavailability, demonstrated enhanced solubility and dissolution when processed using Tween 80 and Aerosil 200 [97]. Characterization techniques such as FTIR and DSC confirmed the transformation of these drugs into amorphous forms, contributing to their improved solubility. Additionally, the technique has shown potential in producing sustained-release formulations and protecting photosensitive drugs from degradation. These findings demonstrate the versatility and scalability of liquisolid technology as a cost-effective and efficient solution for overcoming the limitations of poorly soluble drugs. Another study formulated liquisolid tablets of Pioglitazone HCl to enhance its poor solubility, dissolution rate, and oral bioavailability [98]. Utilizing a liquid-solid technique with optimized carriers like MCC and coating materials like colloidal silicon dioxide, the tablets achieved 99.87% drug release within 60 min. Characterization methods, including FTIR, DSC, and XRD, confirmed improved solubility through the amorphization of the drug without chemical incompatibility. In vivo studies in rabbits demonstrated a 3.06 fold increase in AUC and a 4.18-fold rise in Cmax compared to pure drug, confirming enhanced bioavailability (fig. 3). The approach proved stable under accelerated conditions and offers a cost-effective solution for similar poorly soluble drugs.
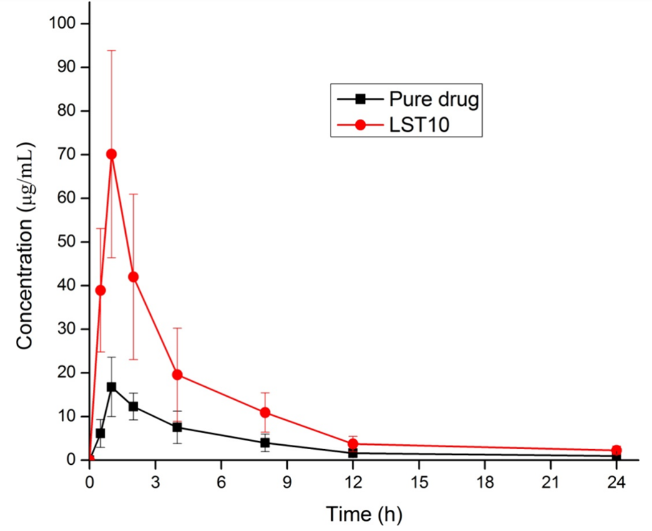
Fig. 3: Plasma concentration profiles of the pure drug pioglitazone HCl and the optimized formulation LST10 (mean±SD, n = 3) [98]
Lyophilization/Freeze-drying technique
Freeze-drying, also known as lyophilization, is a technique that removes solvent from a frozen solution through sublimation. This process consists of three main phases: freezing, primary drying (sublimation), and secondary drying (desorption) [99]. While most commercial products use aqueous solutions for lyophilization, hydrophobic and poorly soluble drugs pose challenges for water-based freeze-drying. Consequently, formulations using pure organic solvents or organic solvent-water mixtures have gained attention in recent years. Tert-butanol (TBA), a low-toxicity, FDAaccepted class 3 solvent with a high vapor pressure and melting point (around 24 °C), is an excellent alternative as a freeze-drying medium [100]. The use of nonaqueous solvents offers several advantages, including enhanced drug solubility, faster sublimation rates, improved chemical stability of both the predried solution and the final product, and easier bulk solution preparation due to increased wettability and solubility of the drug. A group of researcher developed a lyophilized third-generation solid dispersion (SD) of Resveratrol (RES) using Eudragit E PO and Gelucire 44/14 to enhance solubility, dissolution, and bioavailability [101]. The SD transformed RES into an amorphous state, achieving an 8-12 fold solubility increase across various pH levels and a 3 fold boost in bioavailability compared to second-generation SDs. Improved intestinal permeability and reduced P-gp efflux further contributed to enhanced RES absorption, highlighting the potential of this approach for maximizing RES’s therapeutic benefits. Moreover, to enhance the physicochemical properties of vardenafil hydrochloride (VAR), its amorphous form and combinations with excipients such as hydroxypropyl methylcellulose (HPMC) and β-cyclodextrin (β-CD) were developed by Wiergowska et al. [102]. Another work aimed to enhance the physicochemical properties and oral bioavailability of quetiapine fumarate (QF) by developing freeze-dried solid dispersions with nicotinamide (NIC) as a highly soluble conformer [103].
Spray drying
Spray drying is a well-established technique widely employed to enhance the solubility and dissolution of drugs with poor water solubility, particularly those classified under BCS classes II and IV [104, 105]. Key process parameters, including solvent selection, feed rate, and inlet/outlet temperatures, are optimized to ensure uniform particle formation while preserving the integrity of the active pharmaceutical ingredient (API). The amorphous state achieved through spray drying significantly enhances the drug's dissolution and bioavailability by improving wettability and dissolution rates [28].
Spray-dried dispersions (SDDs) are a prominent application of spray drying technology, offering a practical solution to solubility challenges. By dispersing the drug at the molecular level within a polymer matrix, SDDs effectively prevent aggregation and crystallization, enabling controlled and consistent drug release [106]. The dissolution process typically follows either a polymer erosion mechanism, where hydrated polymers release the drug progressively, or a phase separation mechanism, forming nanoparticles that sustain supersaturation. Numerous case studies have demonstrated the efficacy of this technology [107]. For instance, famotidine formulated as an SDD with hydroxypropyl-β-cyclodextrin and PVP K-30 achieved a 38-fold increase in solubility, with over 98% of the drug dissolving within 20 min [108]. Similarly, valsartan SDDs prepared with Eudragit and Pluronic F127 achieved complete dissolution in acidic media, far outperforming the less than 2% dissolution observed with the marketed product [109]. Moreover, Patel et al. [110] developed a gastro-retentive spray-dried dispersion (SDD) formulation of Posaconazole using Soluplus® and Gelucire 43/01. A 32 full factorial design optimized variables, resulting in an SDD with 85% floating efficiency, a 38% increase in solubility, and a 95% drug release in 0.1 N HCl. In vivo studies demonstrated 8-hour gastric retention in rabbits and a 1.5-fold increase in bioavailability in rats compared to the marketed Noxafil® formulation under both fasting and fed conditions. These results highlight the significant potential of SDDs for enhancing solubility, modified release, and bioavailability of poorly soluble drugs. The details summary of recently published work on solubility enhancement has been exemplified in table 1.
Table 1: Recent studied based on innovative technologies for solubility enhancement of poorly soluble drugs
| Therapeutic used | Technique/Approach | Key materials/Excipients used | Outcomes | Ref |
| Posaconazole | Spray dried ASD | Soluplus, Gelucire 43/01 | Pharmacokinetic studies in rats demonstrated improved bioavailability under fasting and fed conditions compared to the marketed Noxafil formulation. | [110] |
| Resveratrol | Lyophilized ASD | Eudragit E PO, Gelucire 44/14 | The third-generation SD demonstrated enhanced RES solubilization, achieving 8-, 12-, and 8-fold improvements compared to pure RES at pH levels 1.2, 4.5, and 6.8, respectively. | [101] |
| Ibrutinib | Hot melt extrusion based ASD | Eudragit® FS100, Poloxamer 407, TPGS, poly(2-ethyl-2-oxazoline) | ASD of Ibrutinib (IBR) achieved>96% release at colonic pH in 6 h and enhanced anticancer activity against colon cancer cell lines. This strategy improves solubility and effectively targets colorectal cancer. | [111] |
| Curcumin | Solvent evaporation based ASD | Kollidon CLSF and surfactant (TPGS or HPMC) | Solid dispersions of curcumin with TPGS improved solubility, stability, oral bioavailability, and gastric ulcer healing, offering an effective pharmaceutical strategy. | [112] |
| Rosuvastatincalcium | Fusion method based ASD | PEG 4000 or pluronic F-127 | The developed orodispersible films of Rosuvastatin showed enhanced solubility, fast disintegration, and 99.06% drug release in 10 min. Pharmacokinetic studies in rabbits confirmed improved bioavailability compared to Crestor® tablets. | [113] |
CONCLUSION
The solubility and bioavailability of poorly water-soluble drugs remain pivotal challenges in pharmaceutical development. Traditional methods, while effective for certain compounds, often fall short when addressing the complexities of modern APIs. Advanced techniques such as nanotechnology, cryogenic grinding, spherical crystallization, and micellar solubilization offer transformative potential, enabling higher solubility, improved stability, and controlled release. These methods not only enhance the dissolution rates but also align with sustainability and scalability in drug production. The review underscores the need for an integrated approach, combining traditional and innovative strategies tailored to specific drug characteristics. Future research should focus on refining these technologies and exploring hybrid methodologies to bridge the gap between laboratory-scale innovation and industrial application. This synthesis of solubility enhancement techniques is poised to advance therapeutic efficacy and patient outcomes in pharmaceutical science.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
All authors have contributed equally
CONFLICT OF INTERESTS
Declared none
REFERENCES
Nyamba I, Sombie CB, Yabre M, Zime Diawara H, Yameogo J, Ouedraogo S. Pharmaceutical approaches for enhancing solubility and oral bioavailability of poorly soluble drugs. Eur J Pharm Biopharm. 2024;204:114513. doi: 10.1016/j.ejpb.2024.114513, PMID 39313163.
Chettri A, Subba A, Singh GP, Bag PP. Pharmaceutical co-crystals: a green way to enhance drug stability and solubility for improved therapeutic efficacy. J Pharm Pharmacol. 2024;76(1):1-12. doi: 10.1093/jpp/rgad097, PMID 37934904.
Salunke S, O Brien F, Cheng Thiam Tan DC, Harris D, Math MC, Arien T. Oral drug delivery strategies for development of poorly water soluble drugs in paediatric patient population. Adv Drug Deliv Rev. 2022;190:114507. doi: 10.1016/j.addr.2022.114507, PMID 36049580.
Ueda K, Moseson DE, Taylor LS. Amorphous solubility advantage: theoretical considerations, experimental methods and contemporary relevance. J Pharm Sci. 2025;114(1):18-39. doi: 10.1016/j.xphs.2024.08.029, PMID 39222748.
Buya AB, Beloqui A, Memvanga PB, Preat V. Self-nano-emulsifying drug delivery systems: from the development to the current applications and challenges in oral drug delivery. Pharmaceutics. 2020;12(12):1194. doi: 10.3390/pharmaceutics12121194, PMID 33317067.
Khan KU, Minhas MU, Badshah SF, Suhail M, Ahmad A, Ijaz S. Overview of nanoparticulate strategies for solubility enhancement of poorly soluble drugs. Life Sci. 2022;291:120301. doi: 10.1016/j.lfs.2022.120301, PMID 34999114.
Das B, Baidya AT, Mathew AT, Yadav AK, Kumar R. Structural modification aimed for improving solubility of lead compounds in early phase drug discovery. Bioorg Med Chem. 2022;56:116614. doi: 10.1016/j.bmc.2022.116614, PMID 35033884.
Khadka P, Ro J, Kim H, Kim I, Kim JT, Kim H. Pharmaceutical particle technologies: an approach to improve drug solubility, dissolution and bioavailability. Asian J Pharm Sci. 2014;9(6):304-16. doi: 10.1016/j.ajps.2014.05.005.
Chaudhary N, Tripathi D, Rai AK. A technical approach of solubility enhancement of poorly soluble drugs: liquisolid technique. Curr Drug Deliv. 2020;17(8):638-50. doi: 10.2174/1567201817666200516155733, PMID 32416691.
Xie B, Liu Y, Li X, Yang P, He W. Solubilization techniques used for poorly water-soluble drugs. Acta Pharmacol Sin B. 2024;14(11):4683-716. doi: 10.1016/j.apsb.2024.08.027, PMID 39664427.
Csicsak D, Szollath R, Kadar S, Ambrus R, Bartos C, Balogh E. The effect of the particle size reduction on the biorelevant solubility and dissolution of poorly soluble drugs with different acid base character. Pharmaceutics. 2023;15(1):278. doi: 10.3390/pharmaceutics15010278, PMID 36678907.
Nykamp G, Carstensen U, Muller BW. Jet milling a new technique for microparticle preparation. Int J Pharm. 2002;242(1-2):79-86. doi: 10.1016/S0378-5173(02)00150-3, PMID 12176228.
Shariare MH, Blagden N, De De Matas M, Leusen FJ, York P. Influence of solvent on the morphology and subsequent comminution of ibuprofen crystals by air jet milling. J Pharm Sci. 2012;101(3):1108-19. doi: 10.1002/jps.23003, PMID 22161641.
Shariare MH, De Matas M, York P. Effect of crystallisation conditions and feedstock morphology on the aerosolization performance of micronised salbutamol sulphate. Int J Pharm. 2011;415(1-2):62-72. doi: 10.1016/j.ijpharm.2011.05.043, PMID 21683128.
Loh ZH, Samanta AK, Sia Heng PW. Overview of milling techniques for improving the solubility of poorly water soluble drugs. Asian J Pharm Sci. 2015;10(4):255-74. doi: 10.1016/j.ajps.2014.12.006.
Vogt M, Vertzoni M, Kunath K, Reppas C, Dressman JB. Cogrinding enhances the oral bioavailability of EMD 57033, a poorly water soluble drug in dogs. Eur J Pharm Biopharm. 2008;68(2):338-45. doi: 10.1016/j.ejpb.2007.06.011, PMID 17646091.
Llorente A, Serrano B, Baselga J, Gedler G, Ozisik R. Jet milling as an alternative processing technique for preparing polysulfone hard nanocomposites. Adv Mater Sci Eng. 2019;2019:1-8. doi: 10.1155/2019/3501402.
Djokic M, Djuris J, Solomun L, Kachrimanis K, Djuric Z, Ibric S. The influence of spiral jet milling on the physicochemical properties of carbamazepine form III crystals: quality by design approach. Chem Eng Res Des. 2014;92(3):500-8. doi: 10.1016/j.cherd.2013.09.011.
Martinez LM, Cruz Angeles J, Vazquez Davila M, Martinez E, Cabada P, Navarrete Bernal C. Mechanical activation by ball milling as a strategy to prepare highly soluble pharmaceutical formulations in the form of co-amorphous co-crystals or polymorphs. Pharmaceutics. 2022;14(10):2003. doi: 10.3390/pharmaceutics14102003, PMID 36297439.
Czajkowska Kosnik A, Misztalewska Turkowicz I, Wilczewska AZ, Basa A, Winnicka K. Solid dispersions obtained by ball milling as delivery platform of etodolac a model poorly soluble drug. Materials (Basel). 2024;17(16):3923. doi: 10.3390/ma17163923, PMID 39203102.
Homayouni A, Sohrabi M, Amini M, Varshosaz J, Nokhodchi A. Effect of high pressure homogenization on physicochemical properties of curcumin nanoparticles prepared by antisolvent crystallization using HPMC or PVP. Mater Sci Eng C Mater Biol Appl. 2019;98:185-96. doi: 10.1016/j.msec.2018.12.128, PMID 30813018.
Karmwar P, Graeser K, Gordon KC, Strachan CJ, Rades T. Effect of different preparation methods on the dissolution behaviour of amorphous indomethacin. Eur J Pharm Biopharm. 2012;80(2):459-64. doi: 10.1016/j.ejpb.2011.10.006, PMID 22019529.
Karmwar P, Graeser K, Gordon KC, Strachan CJ, Rades T. Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods. Int J Pharm. 2011;417(1-2):94-100. doi: 10.1016/j.ijpharm.2010.12.019, PMID 21182910.
Wojnarowska Z, Grzybowska K, Adrjanowicz K, Kaminski K, Paluch M, Hawelek L. Study of the amorphous glibenclamide drug: analysis of the molecular dynamics of quenched and cryomilled material. Mol Pharm. 2010;7(5):1692-707. doi: 10.1021/mp100077c, PMID 20669906.
Bhatia M, Devi S. Co-crystallization: a green approach for the solubility enhancement of poorly soluble drugs. CrystEngComm. 2024;26(3):293-311. doi: 10.1039/D3CE01047C.
Eesam S, Bhandaru JS, Naliganti C, Bobbala RK, Akkinepally RR. Solubility enhancement of carvedilol using drug–drug cocrystallization with hydrochlorothiazide. Futur J Pharm Sci. 2020;6(1):77. doi: 10.1186/s43094-020-00083-5.
Skrdla PJ, Floyd PD, Dell Orco PC. Predicting the solubility enhancement of amorphous drugs and related phenomena using basic thermodynamic principles and semi-empirical kinetic models. Int J Pharm. 2019;567:118465. doi: 10.1016/j.ijpharm.2019.118465, PMID 31279056.
Bhujbal SV, Mitra B, Jain U, Gong Y, Agrawal A, Karki S. Pharmaceutical amorphous solid dispersion: a review of manufacturing strategies. Acta Pharmacol Sin B. 2021;11(8):2505-36. doi: 10.1016/j.apsb.2021.05.014, PMID 34522596.
Kumari L, Choudhari Y, Patel P, Gupta GD, Singh D, Rosenholm JM. Advancement in solubilization approaches: a step towards bioavailability enhancement of poorly soluble drugs. Life (Basel). 2023;13(5):1099. doi: 10.3390/life13051099, PMID 37240744.
Taniguchi C, Kawabata Y, Wada K, Yamada S, Onoue S. Microenvironmental pH-modification to improve dissolution behavior and oral absorption for drugs with pH-dependent solubility. Expert Opin Drug Deliv. 2014;11(4):505-16. doi: 10.1517/17425247.2014.881798, PMID 24472170.
Tran TT, Tran PH, Choi HG, Han HK, Lee BJ. The roles of acidifiers in solid dispersions and physical mixtures. Int J Pharm. 2010;384(1-2):60-6. doi: 10.1016/j.ijpharm.2009.09.039, PMID 19782736.
Almotairy A, Almutairi M, Althobaiti A, Alyahya M, Sarabu S, Alzahrani A. Effect of pH modifiers on the solubility dissolution rate and stability of telmisartan solid dispersions produced by hot-melt extrusion technology. J Drug Deliv Sci Technol. 2021;65:102674. doi: 10.1016/j.jddst.2021.102674, PMID 34552669.
Wu H, Ma J, Qian S, Jiang W, Liu Y, Li J. Co-amorphization of posaconazole using citric acid as an acidifier and a co-former for solubility improvement. J Drug Deliv Sci Technol. 2023;80:104136. doi: 10.1016/j.jddst.2022.104136.
Kumar V, Bharate SB, Vishwakarma RA, Bharate SS. Selection of a water soluble salt form of a preclinical candidate IIIM-290: multiwall plate salt screening and characterization. ACS Omega. 2018;3(7):8365-77. doi: 10.1021/acsomega.8b00801, PMID 30087943.
Serajuddin AT. Salt formation to improve drug solubility. Adv Drug Deliv Rev. 2007;59(7):603-16. doi: 10.1016/j.addr.2007.05.010, PMID 17619064.
Gupta D, Bhatia D, Dave V, Sutariya V, Varghese Gupta S. Salts of therapeutic agents: chemical physicochemical and biological considerations. Molecules. 2018;23(7):1719. doi: 10.3390/molecules23071719, PMID 30011904.
Dwichandra Putra O, Umeda D, Fujita E, Haraguchi T, Uchida T, Yonemochi E. Solubility improvement of benexate through salt formation using artificial sweetener. Pharmaceutics. 2018;10(2):64. doi: 10.3390/pharmaceutics10020064, PMID 29861459.
Choudhury H, Gorain B, Madheswaran T, Pandey M, Kesharwani P, Tekade RK. Drug complexation. In: dosage form design considerations. Amsterdam: Elsevier; 2018. p. 473-512. doi: 10.1016/B978-0-12-814423-7.00014-9.
Kali G, Haddadzadegan S, Bernkop Schnurch A. Cyclodextrins and derivatives in drug delivery: new developments relevant clinical trials and advanced products. Carbohydr Polym. 2024;324:121500. doi: 10.1016/j.carbpol.2023.121500, PMID 37985088.
Loh GO, Tan YT, Peh KK. Enhancement of norfloxacin solubility via inclusion complexation with β-cyclodextrin and its derivative hydroxypropyl-β-cyclodextrin. Asian J Pharm Sci. 2016;11(4):536-46. doi: 10.1016/j.ajps.2016.02.009.
Yallapu MM, Jaggi M, Chauhan SC. β-Cyclodextrin curcumin self-assembly enhances curcumin delivery in prostate cancer cells. Colloids Surf B Biointerfaces. 2010;79(1):113-25. doi: 10.1016/j.colsurfb.2010.03.039, PMID 20456930.
Pandi P, Bulusu R, Kommineni N, Khan W, Singh M. Amorphous solid dispersions: an update for preparation characterization mechanism on bioavailability stability regulatory considerations and marketed products. Int J Pharm. 2020;586:119560. doi: 10.1016/j.ijpharm.2020.119560, PMID 32565285.
Zu Y, Wu W, Zhao X, Li Y, Zhong C, Zhang Y. The high water solubility of inclusion complex of taxifolin-γ-CD prepared and characterized by the emulsion solvent evaporation and the freeze drying combination method. Int J Pharm. 2014;477(1-2):148-58. doi: 10.1016/j.ijpharm.2014.10.027, PMID 25455767.
Sid D, Baitiche M, Elbahri Z, Djerboua F, Boutahala M, Bouaziz Z. Solubility enhancement of mefenamic acid by inclusion complex with β-cyclodextrin: in silico modelling formulation characterisation and in vitro studies. J Enzyme Inhib Med Chem. 2021;36(1):605-17. doi: 10.1080/14756366.2020.1869225, PMID 33557644.
Rezaee F, Ghoreishi SM, Saadati Ardestani N. Precipitation of advanced nanomedicines (curcumin) using supercritical processing; experimental study design and optimizing operating conditions. J Drug Deliv Sci Technol. 2024;99:105989. doi: 10.1016/j.jddst.2024.105989.
Sharma A, Singh M, Sharma V, Vashishth A, Raj M, Upadhyay SK. Current paradigms in employing self-assembled structures: drug delivery implications with improved therapeutic potential. Colloids Surf B Biointerfaces. 2024;234:113745. doi: 10.1016/j.colsurfb.2024.113745, PMID 38241890.
El Hamd MA, Obaydo RH, Nashed D, El Maghrabey M, Lotfy HM. Hydrotropy and co-solvency: sustainable strategies for enhancing solubility of poorly soluble pharmaceutical active ingredients. Talanta Open. 2025 Aug;11:100391. doi: 10.1016/j.talo.2024.100391.
Kankala RK, Zhang YS, Wang SB, Lee CH, Chen AZ. Supercritical fluid technology: an emphasis on drug delivery and related biomedical applications. Adv Healthc Mater. 2017 Aug;6(16):201700433. doi: 10.1002/adhm.201700433, PMID 28752598.
Tran P, Park JS. Application of supercritical fluid technology for solid dispersion to enhance solubility and bioavailability of poorly water soluble drugs. Int J Pharm. 2021;610:121247. doi: 10.1016/j.ijpharm.2021.121247, PMID 34740762.
Wang X, He S, Wang K, Wang X, Yan T, Yan T. Fabrication of betamethasone micro and nanoparticles using supercritical antisolvent technology: in vitro drug release study and Caco-2 cell cytotoxicity evaluation. Eur J Pharm Sci. 2023;181:106341. doi: 10.1016/j.ejps.2022.106341, PMID 36435356.
Misra SK, Pathak K. Supercritical fluid technology for solubilization of poorly water soluble drugs via micro and naonosized particle generation. ADMET DMPK. 2020;8(4):355-74. doi: 10.5599/admet.811, PMID 35300190.
O Sullivan A, Long B, Verma V, Ryan KM, Padrela L. Solid state and particle size control of pharmaceutical cocrystals using atomization based techniques. Int J Pharm. 2022;621:121798. doi: 10.1016/j.ijpharm.2022.121798, PMID 35525471.
Cocero MJ, Martin A, Mattea F, Varona S. Encapsulation and co-precipitation processes with supercritical fluids: fundamentals and applications. J Supercrit Fluids. 2009;47(3):546-55. doi: 10.1016/j.supflu.2008.08.015.
Seedher N, Kanojia M. Micellar solubilization of some poorly soluble antidiabetic drugs: a technical note. AAPS PharmSciTech. 2008;9(2):431-6. doi: 10.1208/s12249-008-9057-5, PMID 18431666.
Mazzotta E, Chieffallo M, Muzzalupo R, Spingola M, Caputo P, Romeo M. Formulation of polymeric micelles to increase the solubility and photostability of caffeic acid. Molecules. 2024;29(14):3329. doi: 10.3390/molecules29143329, PMID 39064907.
Italiya KS, Basak M, Mazumdar S, Sahel DK, Shrivastava R, Chitkara D. Scalable self-assembling micellar system for enhanced oral bioavailability and efficacy of lisofylline for treatment of type-I diabetes. Mol Pharm. 2019;16(12):4954-67. doi: 10.1021/acs.molpharmaceut.9b00833, PMID 31647676.
Shimizu S, Matubayasi N. The origin of cooperative solubilisation by hydrotropes. Phys Chem Chem Phys. 2016;18(36):25621-8. doi: 10.1039/C6CP04823D, PMID 27711657.
Silva SS, Abranches DO, Pinto AS, Soares BP, Passos H, Ferreira AM. Solubility enhancement of hydrophobic compounds in aqueous solutions using biobased solvents as hydrotropes. Ind Eng Chem Res. 2023;62(30):12021-8. doi: 10.1021/acs.iecr.3c01469.
Boyd BJ, Bergström CA, Vinarov Z, Kuentz M, Brouwers J, Augustijns P. Successful oral delivery of poorly water-soluble drugs both depends on the intraluminal behavior of drugs and of appropriate advanced drug delivery systems. Eur J Pharm Sci. 2019;137:104967. doi: 10.1016/j.ejps.2019.104967, PMID 31252052.
Tran TT, Tran KA, Tran PH. Modulation of particle size and molecular interactions by sonoprecipitation method for enhancing dissolution rate of poorly water soluble drug. Ultrason Sonochem. 2015;24:256-63. doi: 10.1016/j.ultsonch.2014.11.020, PMID 25500098.
Dhumal RS, Biradar SV, Yamamura S, Paradkar AR, York P. Preparation of amorphous cefuroxime axetil nanoparticles by sonoprecipitation for enhancement of bioavailability. Eur J Pharm Biopharm. 2008;70(1):109-15. doi: 10.1016/j.ejpb.2008.04.001, PMID 18502628.
Wang X, Majzoobi M, Farahnaky A. Ultrasound-assisted modification of functional properties and biological activity of biopolymers: a review. Ultrason Sonochem. 2020;65:105057. doi: 10.1016/j.ultsonch.2020.105057, PMID 32172150.
Mazumder S, Dewangan AK, Pavurala N. Enhanced dissolution of poorly soluble antiviral drugs from nanoparticles of cellulose acetate based solid dispersion matrices. Asian J Pharm Sci. 2017;12(6):532-41. doi: 10.1016/j.ajps.2017.07.002, PMID 32104366.
Arabestani MR, Bigham A, Kamarehei F, Dini M, Gorjikhah F, Shariati A. Solid lipid nanoparticles and their application in the treatment of bacterial infectious diseases. Biomed Pharmacother. 2024;174:116433. doi: 10.1016/j.biopha.2024.116433, PMID 38508079.
Azevedo C, Macedo MH, Sarmento B. Strategies for the enhanced intracellular delivery of nanomaterials. Drug Discov Today. 2018;23(5):944-59. doi: 10.1016/j.drudis.2017.08.011, PMID 28919437.
Zhang Z, Lu Y, Qi J, Wu W. An update on oral drug delivery via intestinal lymphatic transport. Acta Pharmacol Sin B. 2021;11(8):2449-68. doi: 10.1016/j.apsb.2020.12.022, PMID 34522594.
Chalikwar SS, Belgamwar VS, Talele VR, Surana SJ, Patil MU. Formulation and evaluation of nimodipine loaded solid lipid nanoparticles delivered via lymphatic transport system. Colloids Surf B Biointerfaces. 2012;97:109-16. doi: 10.1016/j.colsurfb.2012.04.027, PMID 22609590.
Kumar VV, Chandrasekar D, Ramakrishna S, Kishan V, Rao YM, Diwan PV. Development and evaluation of nitrendipine loaded solid lipid nanoparticles: influence of wax and glyceride lipids on plasma pharmacokinetics. Int J Pharm. 2007;335(1-2):167-75. doi: 10.1016/j.ijpharm.2006.11.004, PMID 17161566.
Wilson RJ, Li Y, Yang G, Zhao CX. Nanoemulsions for drug delivery. Particuology. 2022;64:85-97. doi: 10.1016/j.partic.2021.05.009.
Mushtaq A, Mohd Wani S, Malik AR, Gull A, Ramniwas S, Ahmad Nayik G. Recent insights into nanoemulsions: their preparation properties and applications. Food Chem X. 2023;18:100684. doi: 10.1016/j.fochx.2023.100684, PMID 37131847.
Van Jaarsveld E, Du Plessis J, Du Preez JL, Shahzad Y, Gerber M. Formulation and characterisation of artemether loaded nano-emulsion for topical applications. J Drug Deliv Sci Technol. 2022;73:103449. doi: 10.1016/j.jddst.2022.103449.
Wooster TJ, Golding M, Sanguansri P. Impact of oil type on nanoemulsion formation and Ostwald ripening stability. Langmuir. 2008;24(22):12758-65. doi: 10.1021/la801685v, PMID 18850732.
Ali HH, Hussein AA. Oral nanoemulsions of candesartan cilexetil: formulation characterization and in vitro drug release studies. AAPS Open. 2017;3(1):4. doi: 10.1186/s41120-017-0016-7.
Hormann K, Zimmer A. Drug delivery and drug targeting with parenteral lipid nanoemulsions a review. J Control Release. 2016;223:85-98. doi: 10.1016/j.jconrel.2015.12.016, PMID 26699427.
Choradiya BR, Patil SB. A comprehensive review on nanoemulsion as an ophthalmic drug delivery system. J Mol Liq. 2021;339:116751. doi: 10.1016/j.molliq.2021.116751.
Thapa R, Sai K, Saha D, Kushwaha D, Aswal VK, Ghosh Moulick RG. Synthesis and characterization of a nanoemulsion system for solubility enhancement of poorly water-soluble non-steroidal anti-inflammatory drugs. J Mol Liq. 2021;334:115998. doi: 10.1016/j.molliq.2021.115998.
Zoller K, Laffleur F, Claus V, Knoll P, To D, Bernkop Schnurch A. Development and in vivo evaluation of nanoemulsions for oral delivery of low molecular weight heparin. J Drug Deliv Sci Technol. 2023;86:104686. doi: 10.1016/j.jddst.2023.104686.
He H, Lu Y, Qi J, Zhu Q, Chen Z, Wu W. Adapting liposomes for oral drug delivery. Acta Pharm Sin B. 2019;9(1):36-48. doi: 10.1016/j.apsb.2018.06.005, PMID 30766776.
Fresta M, Villari A, Puglisi G, Cavallaro G. 5-fluorouracil: various kinds of loaded liposomes: encapsulation efficiency storage stability and fusogenic properties. International Journal of Pharmaceutics. 1993;99(2-3):145-56. doi: 10.1016/0378-5173(93)90356-K.
Eugster R, Luciani P. Liposomes: bridging the gap from lab to pharmaceuticals. Curr Opin Colloid Interface Sci. 2025;75:101875. doi: 10.1016/j.cocis.2024.101875.
Loew S, Fahr A, May S. Modeling the release kinetics of poorly water soluble drug molecules from liposomal nanocarriers. J Drug Deliv. 2011;2011:376548. doi: 10.1155/2011/376548, PMID 21773045.
Aloisio C, Antimisiaris SG, Longhi MR. Liposomes containing cyclodextrins or meglumine to solubilize and improve the bioavailability of poorly soluble drugs. J Mol Liq. 2017;229:106-13. doi: 10.1016/j.molliq.2016.12.035.
Sharma VM, Valsaraj TV, Venkataramana Sudeep H, Raj A, Kodimule S, Jacob J. Preparation characterization in vitro and in vivo studies of liposomal berberine using novel natural fiber interlaced liposomal technology. Eur J Pharm Biopharm. 2024;203:114431. doi: 10.1016/j.ejpb.2024.114431, PMID 39094668.
Kajdic S, Zupancic S, Roskar R, Kocbek P. The potential of nanofibers to increase solubility and dissolution rate of the poorly soluble and chemically unstable drug lovastatin. Int J Pharm. 2020;573:118809. doi: 10.1016/j.ijpharm.2019.118809, PMID 31678525.
Abdulhussain R, Adebisi A, Conway BR, Asare Addo K. Electrospun nanofibers: exploring process parameters polymer selection and recent applications in pharmaceuticals and drug delivery. J Drug Deliv Sci Technol. 2023;90:105156. doi: 10.1016/j.jddst.2023.105156.
Shafi H, Reddy DV, Rashid R, Roy T, Kawoosa S, Bader GN. Optimizing the fabrication of electrospun nanofibers of prochlorperazine for enhanced dissolution and permeation properties. Biomater Adv. 2024;158:213773. doi: 10.1016/j.bioadv.2024.213773, PMID 38277903.
Hartini N, Wu YS, Kusumaningtyas RD, Sriariyanun M, Wu JJ, Cheng YS. Solubility enhancement of curcumin via fast dissolving electrospun nanofibrous mats comprising jelly fig polysaccharides and pullulan. J Taiwan Inst Chem Eng. 2024;160:105346. doi: 10.1016/j.jtice.2024.105346.
Gia Thien Ho T, Huynh TK, Le TK, Nguyen LH, Ton AK, Phan NK. Fabrication and characterization of electrospun diosmetin loaded membranes for enhanced solubility. Chemistry Select. 2024;9(19):e202400633. doi: 10.1002/slct.202400633.
Begum SK, Varma MM, Raju DB, Prasad RG, Phani AR, Jacob B. Enhancement of dissolution rate of piroxicam by electrospinning technique. Adv Nat Sci: Nanosci Nanotechnol. 2012;3(4):45012. doi: 10.1088/2043-6262/3/4/045012.
Xing X, Ouyang J, Guo S, Chen M, Gao Z, He F. Spherical particles design of vanillin via crystallization method: preparation characterization and mechanism. Sep Purif Technol. 2023 Jun 1;314:123622. doi: 10.1016/j.seppur.2023.123622.
Pitt K, Pena R, Tew JD, Pal K, Smith R, Nagy ZK. Particle design via spherical agglomeration: a critical review of controlling parameters rate processes and modelling. Powder Technol. 2018;326:327-43. doi: 10.1016/j.powtec.2017.11.052.
Honmane S, Kadam A, Choudhari S, Patil R, Ansari SA, Gaikwad V. Effect of polymers and process parameters in augmenting the compactability and dissolution behaviour of oxcarbazepine spherical agglomerates. J Drug Deliv Sci Technol. 2021;64:102578. doi: 10.1016/j.jddst.2021.102578.
Varia U, Patel A, Katariya H, Detholia K. Formulation and optimization of polymeric agglomerates of bosentan monohydrate by crystallo-co-agglomeration technique. Bull Natl Res Cent. 2022;46(1):156. doi: 10.1186/s42269-022-00837-6.
Hussain Y, Cui J, Dormocara A, Khan H. The most recent advances in liquisolid technology: perspectives in the pharmaceutical industry. Pharmaceutical Science Advances. 2024 Dec;2:100038. doi: 10.1016/j.pscia.2024.100038.
Lu M, Xing H, Jiang J, Chen X, Yang T, Wang D. Liquisolid technique and its applications in pharmaceutics. Asian J Pharm Sci. 2017;12(2):115-23. doi: 10.1016/j.ajps.2016.09.007, PMID 32104320.
Prajapat MD, Butani SB, Gohel MC. Liquisolid: a promising technique to improve dissolution efficiency and bioavailability of poorly water soluble nimodipine. J Drug Deliv Sci Technol. 2019;53:101135. doi: 10.1016/j.jddst.2019.101135.
Aghajanpour S, Yousefi Jordehi S, Farmoudeh A, Negarandeh R, Lam M, Ebrahimnejad P. Applying liquisolid technique to enhance curcumin solubility: a central composite design study. Chem Pap. 2024;78(17):9257-71. doi: 10.1007/s11696-024-03741-7.
Swain RP, Elhassan GO, Bhattacharjee A, Sahu RK, Khan J. Improved dissolution time and oral bioavailability of pioglitazone using liquisolid tablets: formulation in vitro characterization and in vivo pharmacokinetics in rabbits. ACS Omega. 2024;9(42):42687-97. doi: 10.1021/acsomega.3c09145, PMID 39464449.
Pardeshi SR, Deshmukh NS, Telange DR, Nangare SN, Sonar YY, Lakade SH. Process development and quality attributes for the freeze drying process in pharmaceuticals biopharmaceuticals and nanomedicine delivery: a state of the art review. Futur J Pharm Sci. 2023;9(1):99. doi: 10.1186/s43094-023-00551-8.
Ni N, Tesconi M, Tabibi SE, Gupta S, Yalkowsky SH. Use of pure t-butanol as a solvent for freeze drying: a case study. Int J Pharm. 2001;226(1-2):39-46. doi: 10.1016/S0378-5173(01)00757-8, PMID 11532568.
Almeida H, Ferreira B, Fernandes Lopes C, Araujo F, Bonifacio MJ, Vasconcelos T. Third generation solid dispersion through lyophilization enhanced oral bioavailability of resveratrol. ACS Pharmacol Transl Sci. 2024;7(3):888-98. doi: 10.1021/acsptsci.4c00029, PMID 38481698.
Wiergowska G, Ludowicz D, Wdowiak K, Miklaszewski A, Lewandowska K, Cielecka Piontek J. Combinations of freeze dried amorphous vardenafil hydrochloride with saccharides as a way to enhance dissolution rate and permeability. Pharmaceuticals (Basel). 2021;14(5):453. doi: 10.3390/ph14050453, PMID 34064796.
Ali AM, Al Remawi MM. Freeze dried quetiapine nicotinamide binary solid dispersions: a new strategy for improving physicochemical properties and ex vivo diffusion. J Pharm (Cairo). 2016;2016:2126056. doi: 10.1155/2016/2126056, PMID 28042494.
Patil A, Patil P, Pardeshi S, Shrimal P, Rebello N, Mohite PB. Combined microfluidics and drying processes for the continuous production of micro-/nanoparticles for drug delivery: a review. Drying Technology. 2023;41(10):1533-68. doi: 10.1080/07373937.2023.2167827.
Pardeshi S, More M, Patil P, Pardeshi C, Deshmukh P, Mujumdar A. A meticulous overview on drying based (spray, freeze, and spray-freeze) particle engineering approaches for pharmaceutical technologies. Drying Technology. 2021;39(11):1447-91. doi: 10.1080/07373937.2021.1893330.
Friesen DT, Shanker R, Crew M, Smithey DT, Curatolo WJ, Nightingale JA. Hydroxypropyl methylcellulose acetate succinate based spray-dried dispersions: an overview. Mol Pharm. 2008;5(6):1003-19. doi: 10.1021/mp8000793, PMID 19040386.
Vodak DT, Morgen M. Design and development of HPMCAS-based spray dried dispersions. Adv Deliv Sci Technol. 2014:303-22. doi: 10.1007/978-1-4939-1598-9_9.
Verma U, Naik JB, Patil JS, Yadava SK. Screening of process variables to enhance the solubility of famotidine with 2-HydroxyPropyl–β-Cyclodextrin & PVP K-30 by using plackett–burman design approach. Mater Sci Eng C. 2017;77:282-92. doi: 10.1016/j.msec.2017.03.238.
Pradhan R, Kim SY, Yong CS, Kim JO. Preparation and characterization of spray dried valsartan loaded eudragit® E PO solid dispersion microparticles. Asian J Pharm Sci. 2016;11(6):744-50. doi: 10.1016/j.ajps.2016.05.002.
Patel K, Kevlani V, Shah S. A novel posaconazole oral formulation using spray dried solid dispersion technology: in vitro and in vivo study. Drug Deliv Transl Res. 2024;14(5):1253-76. doi: 10.1007/s13346-023-01461-1, PMID 37952081.
Patel H, Palekar S, Patel A, Patel K. Ibrutinib amorphous solid dispersions with enhanced dissolution at colonic pH for the localized treatment of colorectal cancer. Int J Pharm. 2023;641:123056. doi: 10.1016/j.ijpharm.2023.123056, PMID 37207861.
Xi Z, Fei Y, Wang Y, Lin Q, Ke Q, Feng G. Solubility improvement of curcumin by crystallization inhibition from polymeric surfactants in amorphous solid dispersions. J Drug Deliv Sci Technol. 2023 May;83:104351. doi: 10.1016/j.jddst.2023.104351.
Zaki RM, Alfadhel M, DevanathaDesikan Seshadri V, Albagami F, Alrobaian M, Tawati SM. Fabrication and characterization of orodispersible films loaded with solid dispersion to enhance rosuvastatin calcium bioavailability. Saudi Pharm J. 2023;31(1):135-46. doi: 10.1016/j.jsps.2022.11.012, PMID 36685296.