
Int J Pharm Pharm Sci, Vol 17, Issue 5, 8-16Original Article
PREPARATION AND EVALUATION OF RABEPRAZOLE SODIUM COMPRESSION-COATED TABLETS FOR CHRONOTHERAPY
SARFARAZ MD*, VIJAY ARCHAK, H. DODDAYYA
Department of Pharmaceutics, NET Pharmacy College, Raichur-584103, Karnataka, India
*Corresponding author: Sarfaraz Md; *Email: sarfindia@gmail.com
Received: 06 Dec 2024, Revised and Accepted: 17 Mar 2025
ABSTRACT
Objective: The objective of research work was to prepare and evaluate rabeprazole sodium compression-coated tablets designed for chronotherapy in treating acidity-related disorders, such as peptic ulcer disease and Gastroesophageal Reflux Disease (GERD).
Methods: Direct compression method was used to prepare the compression-coated tablets. The formulation process utilized various super disintegrants for core tablets and polymers to create coated tablets that exhibit a pulsatile release profile, ensuring rapid drug release post-lag time. Comprehensive evaluations of the physical properties, hardness, friability, weight variation, drug content, swelling index, disintegration time and in vitro drug release studies were done for the tablets.
Results: The findings indicated that the developed compression-coated tablets demonstrated optimal mechanical stability, uniformity in drug content, and a desirable release pattern. The optimised formulation C5 achieved 99.85% drug release within 15 min after 4.15 h of lag time. Kinetic studies revealed a first-order release mechanism, and compatibility assessments via Fourier Transformer Infrared Spectrophotometer (FTIR) confirmed the stability of rabeprazole in the presence of the excipients.
Conclusion: The study highlights the significance of chronomodulated drug delivery systems (CDDS) that can delay drug release for a predetermined duration, aligning with the circadian rhythm of gastrointestinal function. This research provides a promising approach to enhance the therapeutic effectiveness of rabeprazole through a chronotherapeutic strategy developed using compression coating, potentially improving patient outcomes in managing gastric acid-related conditions.
Keywords: Rabeprazole sodium, Sodium starch glycolate, Crospovidone, Croscarmellose sodium, HPMC, HPC, Direct compression method
© 2025 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijpps.2025v17i5.53353 Journal homepage: https://innovareacademics.in/journals/index.php/ijpps
INTRODUCTION
The evolution of drug delivery systems has transitioned from traditional methods of simple absorption to advanced mechanisms that align with biological rhythms, epitomized by the emergence of chronotherapeutics. Chronomodulated drug delivery systems with innovative designs are engineered to provide rapid and transient release of therapeutic agents in response to specific biological demands. Unlike conventional continuous delivery methods, CDDS are tailored for diseases that require intermittent therapeutic concentrations rather than a steady-state drug level. By synchronizing drug release with the physiological needs of patients, this approach aims to enhance therapeutic efficacy and patient compliance, thereby representing a significant advancement in the field of pharmacotherapy. These systems underscore the importance of matching drug delivery timing with disease pathology, paving the way for future research and development in the domain of chronopharmacology [1]. The prevalence and characteristics of nocturnal pain in patients suffering from peptic ulcers and gastroesophageal reflux disease are well known. The symptoms of epigastralgia and retrosternal pain are notably exacerbated during the nighttime hours, particularly from midnight to dawn. In chronotherapeutic diseases, a correlation between the timing of the symptoms and underlying pathophysiological mechanisms exists and there is a significant need for targeted therapeutic interventions that address nighttime symptomatology in these affected patients, paving the way for advancements in personalized medicine and improved patient compliance [2, 3].
Compression coating is a method employed to create specialized modified-release pharmaceutical products. Also known as press coating or dry coating, this process involves the application of fine dry granules onto a core drug tablet using a specifically designed tablet press. It is suitable for heat and moisture-sensitive drugs and can produce repeat-action and timed-action tablets, where the entire surface of the core is enveloped by the coating. The coatings serve a crucial role in controlling drug release, ensuring that the active ingredients are only released once the polymeric or drug coat is completely eroded, dissolved, or removed [4]. Disintegrants play a crucial role in the formulation of pharmaceutical tablets and capsules by facilitating the rapid dispersion of active ingredients. The core tablet containing drug and appropriate disintegrants facilitates water penetration into the core upon contact with an aqueous environment that culminates in the explosive release of the drug, providing a rapid and effective release of the drug in the gastrointestinal tract [5]. The most widely used disintegration agents include starch; various starch derivatives, and numerous natural disintegrants, at a standard concentration of 5%. Selecting appropriate disintegrants to optimize the efficacy of pharmaceutical formulations is important in core tablets. The core tablets are coated with hydrophilic swellable polymers like hydroxy propyl methylcellulose (HPMC), methacrylic and acrylic polymers or polyalcohols, which release drug after lag-time depending on thickness of the coat and viscosity grade of the polymer [6, 7]. Rabeprazole sodium is a medication that decreases stomach acid. It is used to treat peptic ulcer disease, gastroesophageal reflux disease, and excess stomach acid production, such as in Zollinger-Ellison syndrome. The dose is 20 mg orally once a day. The half-life of rabeprazole is 1-2 h (in plasma). Hence, the present research work aimed to prepare and evaluate rabeprazole compression-coated tablets for chronotherapy of acidity for effective treatment of peptic ulcer and gastroesophageal reflux disease etc.
MATERIALS AND METHODS
Rabeprazole sodium was obtained as a gift sample from Enal Drugs Pvt. Ltd., Jedimetla-Hyderabad, Telangana. Sodium starch glycolate, Crospovidone and Aerosil were procured from Himedia Laboratories Pvt Ltd, Thane, Mumbai. Croscarmellose sodium was obtained from Microwax Pvt. Ltd., Mumbai. HPMC K100, HPMC K4 and HPMC K15 were purchased from Shin-estu Chemical Co-Ltd., New Delhi. HPC was obtained from Nippon soda Co-Ltd., Chiyoda-Japan. Microcrystalline cellulose and Lactose were purchased from S. D. Fine Chemicals Pvt. Ltd., Mumbai.
Formulation of rabeprazole sodium compression-coated tablet for chronotherapy
Preparation of core tablets by direct compression
The inner core/immediate release tablets containing rabeprazole sodium were prepared by direct compression method using super disintegrants like sodium starch glycolate, cross povidone and croscarmellose sodium in different amounts of 2.5%, 5% and 7.5%. The powder mixtures of rabeprazole sodium, lactose, sodium starch glycolate, crospovidone, croscarmellose sodium and microcrystalline cellulose were dry blended for 15 min and further blended for 5 min following the addition of aerosil and magnesium stearate. The prepared blends were then evaluated for flow properties like angle of repose, bulk density, tapped density, Carr's index and Hausner's ratio and finally compressed using a 10-station Rimek tablet compression machine to obtain tablets of 7.0 mm diameter and weighing 200 mg each. The tablets were packed in aluminium foil and stored in air-tight containers in a desiccator. The formula of core tablets is given in table 1.
Preparation of compression-coated tablets
The compression-coated tablets of rabeprazole sodium (C1-C16) were prepared by compression coating of the optimised core tablet formulation (F6) with outer barrier layers having different compositions of polymers like HPMC K4, HPMC K100, HPMC K15 and hydroxypropyl cellulose (HPC). The formulae of all tablets are given in table 2. The coating polymers HPMC K4M, HPMC K15M, HPMC K100M and HPC were used in different percentages of 43%, 50%, 55% and 60%. During coating, the first half of the respective polymer was placed uniformly in the die cavity (10 mm) and then the optimized core tablet (F6) was placed centrally and the remaining half of the polymer was added above the core tablet to cover it completely. Then the polymer and core tablets were directly compressed to get coated tablets [8, 9].
Precompression evaluation of powder blend of core tablet
Angle of repose (θ)
The fixed funnel method was employed to measure the angle of repose (θ) and it was calculated using the following formula:
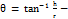 ------- (1)
------- (1)
In which θ is the angle of repose, h is the height of the cone and r is the radius of the cone base. The test was repeated thrice (n = 3).
Bulk density
The bulk density, as a measure used to describe the packing material of the powder, was determined by transferring the accurately weighed powder to the graduated cylinder (10 ml) with the aid of a funnel. The powder was levelled carefully without compacting, and the unsettled volume was read. The volume was noted and bulk density (g/cm3) was determined as the ratio of weight of the sample to the volume occupied. The test was repeated thrice. (n = 3)
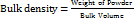 . . (2)
. . (2)
Tapped density
The sample was transferred in to a 10 ml graduated measuring cylinder. Then cylinder was tapped manually by raising the cylinder and allowing it to drop under its own weight. Cylinder was tapped for 100 times and tapped volume was measured. The test was repeated thrice (n = 3). Tapped density in g/cm3was calculated by the following formula:
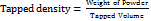 ……… (3)
……… (3)
Table 1: Formula of core tablets
| Ingredient (mg) | F1 | F2 | F3 | F4 | F5 | F6 | F7 | F8 | F9 |
| Rabeprazole sodium | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| Lactose | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 | 70 |
| Sodium starch glycolate | 5 | 10 | 15 | - | - | - | - | - | - |
| Crospovidone | - | - | - | 5 | 10 | 15 | - | - | - |
| Croscarmellose sodium | - | - | - | - | - | - | 5 | 10 | 15 |
| Microcrystalline cellulose | 100 | 95 | 90 | 100 | 95 | 90 | 100 | 95 | 90 |
| Aerosil | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| Magnesium sterate | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| Total | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 |
Table 2: Formula of compression-coated tablets
| Ingredient (mg) | C1 | C2 | C3 | C4 | C5 | C6 | C7 | C8 | C9 | C10 | C11 | C12 | C13 | C14 | C15 | C16 |
| Core tablet | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 | 200 |
| HPMC K4 | 150 | 200 | 250 | 300 | - | - | - | - | - | - | - | - | - | - | - | - |
| HPMC K15 | - | - | - | - | 150 | 200 | 250 | 300 | - | - | - | - | - | - | - | - |
| HPMC K100 | - | - | - | - | - | - | - | - | 150 | 200 | 250 | 300 | - | - | - | - |
| HPC | - | - | - | - | - | - | - | - | - | - | - | - | 150 | 200 | 250 | 300 |
| Total | 350 | 400 | 450 | 500 | 350 | 400 | 450 | 500 | 350 | 400 | 450 | 500 | 350 | 400 | 450 | 500 |
Carr’s index
The flowability of powder samples was also assessed from Carr’s Index (CI). The compressibility of sample blends was determined from their apparent bulk density and the tapped densities by using the following formula:
The test was carried out in triplicate. (n = 3)
 ………… (4)
………… (4)
Hausner’s ratio
Hausner’s ratio is an indication of the flowability of powder and if the ratio is greater than 1.25 is considered to be an indication of poor flowability. Hausner’s ratio was determined by the following equation. The test was done in triplicate (n = 3) [10].
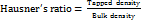 ……………. (5)
……………. (5)
Post compression evaluation
All core and compression-coated tablets were evaluated for various parameters related to physical characteristics, mechanical strength and drug release.
The core tablets were evaluated for weight variation, hardness, friability, disintegration test, drug content, wetting study and in vitro dissolution study. The coated tablets were evaluated for thickness, diameter, hardness, weight variation, friability, drug content, swelling index, in vitro dissolution, kinetic and FTIR studies.
General appearance
Morphological characters like shape, colour and texture were determined visually.
Thickness and diameter
The thickness and diameter of prepared core and compression-coated tablets (10 nos) were tested using Vernier callipers. The test was done in triplicates (n = 3) and the average was determined.
Hardness
The hardness of prepared core and compression-coated tablets (5 nos) was determined by using the Monsanto hardness tester and measured in terms of kg/cm2. The test was done in triplicate (n = 3).
Weight variation
The weight variation test was done for core and compression-coated tablet by weighing 10 tablets (Shimadzu digital balance) individually, calculating the average weight and comparing the individual tablet weights to the average. The percentage difference in the weight variation should be within the permissible limits (±5%). The percentage deviation was calculated using formula (6): The limits as per United State Pharmacopoeia (USP) were followed. The test was done in triplicate (n = 3) [11].
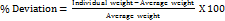 ------ (6)
------ (6)
Friability
The test was performed for core tablets to assess the effect of friction and shocks, which may often cause tablets to chip, cap or break. Roche friabilator was used for testing the friability of prepared tablets. 10 tablets were accurately weighed and placed in the friabilator and operated for 100 revolutions. The tablets were de-dusted and reweighed.
Friability (F) was calculated using the following formula:
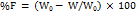 ------------ (7)
------------ (7)
Where W0 is the Initial weight of the tablets and W is the Final weight of tablets, respectively. The test was done in triplicate (n = 3). The tablets that lost less than 1% weight were considered to be compliant [12].
Drug content
The drug content of the core and coated tablet was performed separately. The core and coated tablet (1 No) were powered separately in a glass mortar and pestle. The whole powder of the tablet was taken into a 100 ml volumetric flask and dissolved in pH 6.8 phosphate buffer while shaking for 10 min. Further samples were diluted with buffer to make up the volume to 100 ml. The solution in the volumetric flask was filtered and the drug content was determined at 284 nm by using the UV-spectrophotometer against blank. The test was done in triplicate (n = 3) [13].
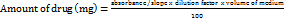 -- (8)
-- (8)
Disintegration time
The disintegration test was carried out in distilled water using the USP disintegration test apparatus from (Electro Lab, ED-2L, Mumbai). Six tablets were placed in the cylindrical glass tubes and the time taken for the tablets to disintegrate was recorded as disintegration time [14, 15]. The test was done in triplicate (n = 3).
Wetting time
This test was done for core tablets. A tissue paper folded twice was placed in the petridish. 10 ml of water was added to the petridish. A tablet was carefully placed on the surface of the tissue paper. The time required for the water to reach upper surface of the tablet was noted as wetting time [16]. The test was done in triplicate (n = 3).
Swelling index
Swelling of coated tablets involves the absorption of a liquid by tablet matrices, resulting in an increase in the weight and volume of the tablet. The extent of swelling was measured in terms of %weight gain by the tablet. For each formulation batch, one tablet was weighed and placed in a petridish containing 10 ml of pH 6.8 phosphate buffer. After each 1 h time interval, the tablet was removed from petridish and weighed again up to 6 h. The test was done in triplicate (n = 3). The swelling index was calculated using the following equation (9) [17].
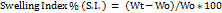 ………. (9)
………. (9)
Where S. I. = Swelling index, Wt = Weight of tablet at time t, Wo = Weight of tablet before placing in the petridish.
In vitro drug release studies
For core tablets
In vitro dissolution studies were conducted for all the core tablets prepared to determine the release pattern of the drug from the tablet. The dissolution test for core tablets was carried out using 8 stations USP Type II dissolution test apparatus (Electro Lab, TDT-O8L, Mumbai). The dissolution studies were carried out in 900 ml pH 6.8 phosphate buffer at 37±0.5 C. The speed of the paddle was set at 50 rpm. Sampling was done every 5 min intervals. An aliquot of 5 ml sample was withdrawn at each time interval and replaced with the equal volume of fresh medium. The samples withdrawn after suitable dilution were analysed in the UV spectrophotometer at 284 nm. The mean of three determinations (n = 3) was used to calculate the drug release from each formulation.
For compression-coated tablets
The dissolution test for compression-coated tablets of rabeprazole was carried out using 8 stations USP Type II dissolution test apparatus (Electro Lab, TDT-O8L, Mumbai). For the first 2 h the dissolution studies were carried out in 900 ml 0.1 N HCl (pH 1.2) and later the medium was replaced with pH 6.8 phosphate buffer till the end of the dissolution study. The speed of the paddle was set at 50 rpm and the temperature was controlled at 37±0.5 ⁰C. Sampling was done every 30 min intervals. An aliquot of 5 ml sample was withdrawn at each time interval and replaced with the equal volume of fresh medium. The samples withdrawn after suitable dilution were analysed in the UV spectrophotometer at 284 nm. The mean of three determinations (n = 3) was used to calculate the drug release from each formulation [18].
Kinetic study
In order to analyze the release mechanism from coated tablets, several release models were tested such as:
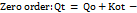 ----------- (10)
----------- (10)
Where Qt is the amount of drug released at time t, Ko is the apparent dissolution rate constant or zero-order release constant and Qo is the initial concentration of the drug in the solution resulting from a burst effect; in this case, the drug release runs as a constant rate.
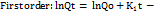 ----------- (11)
----------- (11)
Where K1 is the first-order release constant; in this case, the drug released at each time is proportional to the residual drug inside the dosage form.
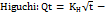 ----------- (12)
----------- (12)
Where Qt is the amount of drug released at time t and KH is the higuchi release rate constant; this is the most widely used model to describe drug release from pharmaceutical matrices.
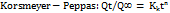 ------------ (13)
------------ (13)
Where Kk is a constant incorporating structural and geometric characteristics of the drug dosage form and n is the release exponent, indicative of the drug release mechanism.
The value of n for a formulation, n = 0.45 for Fickian (Case I) release, >0.45 but<0.89 for non-Fickian (Anomalous) release and 0.89 for Case II (Zero order) release and>0.89 for super case II type of release. Case II transport generally refers to the dissolution of the polymeric matrix due to the relaxation of the polymer chain and anomalous transport (Non-Fickian) refers to the summation of both diffusion and dissolution-controlled release [19].
FTIR studies
The Pure drug and selected formulations (C4, C8, C12, and C16) were subjected for the FTIR analysis to check the compatibility/interaction between the drug and excipients. The samples were prepared on KBr-press (Bruker Alpha FT-IR Spectrometer: ATR MODE, Chitradurga). The samples were scanned over a range of 4000-400 cm-1 using Fourier transformer infrared spectrophotometer. Spectra were analysed for drug carrier interactions [20].
RESULTS AND DISCUSSION
Pre-compression evaluation of powder blend of rabeprazole sodium core tablet
The method employed for the preparation of compression-coated tablets in the study was direct compression for which the drug or the mixture of the drug and polymer should possess good flow properties. Various pre-compression parameters were evaluated for estimating the flow ability of powder. Table 3 shows the flow properties of powder blends. The angle of repose of the powder blend was in the range of 25.5±0.04-30.2±0.06°. As it is below 30° it indicates good flow properties of blend. Bulk density was found between 0.31±0.12-0.38±0.14 gm/cm3 and tapped density between 0.36±0.07-0.42±0.09 gm/cm3for all the formulations. To know the compaction property of powder blends % Carr’s compressibility was calculated. Carr’s index value of 5.10±0.03–12.43±0.08% and Hausner’s ratio of 1.09±0.09-1.18±0.5 were observed for all precompression blends. Carr’s index and Hauser’s ratio of below 15% and 1.25, respectively, are indicative of good compressibility. Hence, powder mixture was found suitable for direct compression.
Post-compression evaluation of core tablets
The core tablets were white, circular and had convex surface on both the sides with sharp edges whereas coated tablet were white coloured circular having flat surfaces on both sides with sharp edges. The results of the post-compression evaluation of core tablets are given in table 4-5. As the material was free-flowing, tablets were obtained of uniform weight due to uniform die-filling. The thickness was found in the range of 4.5±0.1–4.8±0.17 mm. The diameter of tablets was found to be 7±0.00 mm and was uniform for all tablets. The disintegration time of the core tablet was found between 15±0.25–48±0.29 sec. This is due to the fast uptake of the water from the medium, swelling, and burst effect of super disintegrants used in the core tablet. The disintegration time of tablets was inversely proportional to the concentration of super disintegrants. As the amount of super disintegrants increased in the tablet from 2.5-7.5% the disintegration time decreased proportionally from 30 to 15 sec in the case of core tablets with crospovidone. A similar effect was observed with tablets containing sodium starch glycolate and croscarmellose sodium. The tablets containing crospovidone gave lesser disintegration time, which is a desirable property for the core tablets. The performance of superdisintegrants was found in the order of CP>SSG>CCS. The results were similar to study conducted by Mahale Nitin B and Battase AP [21]. The wetting time of the core tablet was found between 25±0.15–96±0.12 sec. On comparing the super disintegrants, the formulations containing crospovidone took less wetting time than the other formulations. Tablets containing crospovidone quickly wicked water and were hydrated; however, were soft as compared with tablets prepared with croscarmellose sodium, sodium starch glycolate. This was similar to the observations of Sanjiv Kumar G et al. [22]. The wetting time of tablets and disintegration time were correlated. Tablets showing less wetting time disintegrated in less time ex: F4, F5, F6. The hardness of tablets was between 2.8±0.22–3.0±0.15 kg/cm2 for all the formulations. The hardness test indicated good mechanical strength of tablets. Friability was found in between 0.97±0.34-0.99±0.34%. The value below 1% was an indication of tablet’s good mechanical resistance. The weight variation of tablets was found within the USP specified limits of ±7.5%. The drug content was found to be in the range of 17.8±0.56-19.8±0.97 mg, which was within the acceptable limits. Some loss in the drug content of core tablet was due to the very light-sensitive nature of the drug molecule.
Table 3: Flow properties of powder blends
| Formulation code | Angle of repose*(θ) | Bulk density* (g/cm3) | Tapped density* (g/cm3) | Carr’s index* (%) | Hausner’s ratio* |
| Pure drug | 26.40±0.80 | 0.33±0.10 | 0.36±0.56 | 8.33±0.04 | 1.09±0.9 |
| F1 | 26.3±0.07 | 0.31±0.12 | 0.41±0.11 | 12.43±0.08 | 1.18±0.5 |
| F2 | 26.5±0.07 | 0.33±0.11 | 0.36±0.07 | 8.33±0.06 | 1.09±0.4 |
| F3 | 27.8±0.08 | 0.37±0.92 | 0.42±0.09 | 11.90±0.04 | 1.13±0.6 |
| F4 | 30.2±0.06 | 0.38±0.14 | 0.40±0.06 | 5.90±0.06 | 1.05±0.8 |
| F5 | 27.8±0.05 | 0.38±0.11 | 0.41±0.04 | 7.31±0.07 | 1.07±0.6 |
| F6 | 25.5±0.04 | 0.37±0.08 | 0.39±0.08 | 5.10±0.03 | 1.05±0.4 |
| F7 | 27.3±0.09 | 0.36±0.15 | 0.38±0.12 | 5.20±0.06 | 1.05±0.9 |
| F8 | 26.9±0.010 | 0.35±0.11 | 0.37±0.13 | 5.40±0.07 | 1.05±0.7 |
| F9 | 29.2±0.012 | 0.38±0.13 | 0.39±0.14 | 5.82±0.08 | 1.02±0.8 |
*Average of 3 determinations,±SD
Table 4: Evaluation of core tablets
| Formulation code | Thickness (mm) | Diameter (mm) | Disintegration (sec) |
| F1 | 4.7± 0.05 | 7±0.00 | 34±0.33 |
| F2 | 4.5±0.1 | 7±0.00 | 24±0.31 |
| F3 | 4.6± 0.03 | 7±0.00 | 18±0.22 |
| F4 | 4.8±0.17 | 7±0.00 | 30±0.22 |
| F5 | 4.5±0.1 | 7±0.00 | 23±0.14 |
| F6 | 4.6±0.03 | 7±0.00 | 15±0.25 |
| F7 | 4.7±0.05 | 7±0.00 | 48±0.29 |
| F8 | 4.8±0.17 | 7±0.00 | 43±0.12 |
| F9 | 4.5±0.1 | 7±0.00 | 38±0.32 |
*Average of 3 determinations,±SD
Table 5: Evaluation of core tablets
| Formulation code | Wetting time (sec) | Hardness test (kg/cm2)* | Friability (%)** | Weight variation (mg)*** | Drug content (mg) |
| F1 | 38±0.18 | 2.8± 0.22 | 0.99±0.21 | 202.3±0.20 | 18.3±0.06 |
| F2 | 32±0.11 | 3.0.±0.15 | 0.98±0.27 | 199.8±0.10 | 18.6±0.24 |
| F3 | 30±0.13 | 3.0±0.12 | 0.99±0.31 | 200.0±0.00 | 19.2±0.84 |
| F4 | 30±0.20 | 2.8±0.18 | 0.98±0.12 | 200.1±0.09 | 18.2±0.16 |
| F5 | 28±0.16 | 2.8±0.22 | 0.95±0.22 | 200.4± 0.04 | 18.8±0.44 |
| F6 | 25±0.15 | 2.8±0.05 | 0.98±0.16 | 200.1± 0.09 | 19.8±0.97 |
| F7 | 96±0.12 | 2.8±0.17 | 0.99±0.34 | 200.0±0.00 | 17.8± 0.56 |
| F8 | 91±0.14 | 2.8±0.19 | 0.98±0.13 | 200.3±0.07 | 19.4±0.92 |
| F9 | 85±0.25 | 2.8±0.17 | 0.97±0.34 | 200.4± 0.04 | 18.8±0.44 |
*All values are expressed as mean±SD, n=5*/10**/20**
Post-compression evaluation of compression-coated tablets
The results of the post-compression evaluation of the compression-coated tablets are given in table 6. The thickness of the coated tablets was found in the range of 3.7±0.73–5.8±0.99 mm. The thickness of coated tablets was proportional to the concentration of polymer in the tablet. The thickness increased as the polymer concentration increased from 43-60% in the coated tablet. The diameter of tablets was found to be 10±0.00 mm and was uniform for all tablets. The hardness of tablets was between 5.1±0.12–5.8±0.22 kg/cm2 for all the formulations. The hardness of tablets with different polymers was adjusted accordingly to proper lag time. The weight of tablets was in the range of 349.4±0.44–500.6±0.44 mg which was within the specified USP limits (±5%). Friability was found in between 0.15±0.18-0.87±0.22%. The value below 1% was an indication of the tablet’s good mechanical resistance. The drug content was found to be 17.48±0.22-19.88±0.22 mg, which was within the acceptable limits. The swelling index (%) of coated tablets was found between 54.2±0.63–98.5±0.57% for all the formulations. The swelling index studies showed a proportional increase with an increase in the concentration of polymers used. HPMC K15 and HPMC K100 exhibited a maximum swelling index of 98.5%, which indicates they swell 2-3 times of their original value. The lowest swelling index was with HPMC K4 of 54.2%. The swelling index for all polymers was in the order HPMC K100>HPMC K15>HPC>HPMC K4. HPMC K100 has a higher molecular weight than other polymer. This higher molecular weight means that HPMC K100 can form a more robust gel layer when it comes into contact with water, leading to faster and more extensive swelling. HPMC K4M has lower molecular weight and viscosities, which results in a slower swelling rate. The gel layer formed by these grades was less dense and took a while to swell.
In vitro drug release studies
Core tablet
The rabeprazole sodium core tablets F1-F3 containing sodium starch glycolate as super disintegrants released 70.20%-91.04% of the drug in 30 min, respectively. Core tablet F4-F6 containing crospovidone as super disintegrants released 74.25%-99.62% of drug in 30 min. Core tablet F7-F9 containing croscarmellose sodium as super disintegrants released 77.31%-90.25% of the drug in 30 min. The maximum drug release was observed with F6 prepared using crospovidone, 99.6% at the end of 30 min. Since F6 released the maximum drug in less amount of time and also has the least disintegration time of 15 sec, it was selected as the optimized core tablet formulation. The order of drug release from all formulations prepared was F6>F5>F3>F9>F8>F2>F7>F4>F1. In case of the different super disintegrants used, the order of release was Crospovidone>sodium starch glycolate>croscarmellose sodium. The observation was consistent with the results obtained by Saripilli R et al. [23]. The results of in vitro release studies are given in fig. 1.
Compression-coated tablets
The release profile of all the compression-coated tablets exhibited a lag time followed by the burst release of the drug. The compression-coated tablets C1-C16 released 98.22–99.85% of the drug in between 4-6 h. The compression-coated tablets comprising HPMC K4M as polymer C1–C4 showed drug release between 98.22-99.85% in 4.25-6 h. The compression-coated tablets comprising HPMC K15M as polymer C5–C8 showed drug release between 98.95–99.85% at the end of 4.20-6 h. Further, the compression-coated tablets composed of HPMC K100M C9–C12 released about 99.11–99.78% of rabeprazole sodium at the end of 4.40-6 h. Formulations C13–C16 composed of HPC as polymer showed a release of about 99.25–99.81% of the drug at the end of 4.35-6.55 h, respectively. The maximum drug release was observed with C5–99.85% at the end of 270 min (4 h 30 min). C5 prepared using HPMC K15M (43%) showed a lag time of 4 h 15 min and burst released the entire drug within 15 min after the lag time. Hence, it is suitable for chronotherapeutic delivery of rabeprazole sodium and, therefore, was selected as an Optimised formulation. The order of drug release from all formulations prepared was C8>C9>C15>C1>C4>C3>C5>C10>C11>C12>C14>C13>C7>C16>C6>C2. In case of the different polymers used, the order of release was HPMC K15>HPMC K100>HPC>HPMC K4. The findings were comparable to Swati CJ et al. [24]. HPC showed a maximum time-controlled release of the drug than other polymers. The results also indicated that the lag time increased as the amount of polymer concentration increased (C1-265 min, C2-315 min, C3-360 min, C4-405 min). The same results were observed with other polymers used. The results of in vitro release studies are given in fig. 2. The swelling studies of HPMC press-coated tablets also showed a proportionate increase in swelling with an increase in the polymer level.
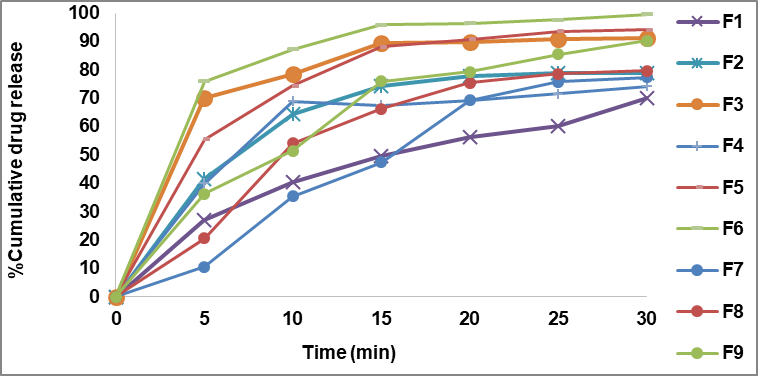
Fig. 1: In vitro release profile of core tablets (F1-F9)
Table 6: Evaluation of compression-coated tabletsᴪ
Formulation code |
Thickness (mm) |
Diameter (mm) |
Hardness Test (kg/cm2)* |
Friability (%)** |
Weight variation (mg) *** |
Drug content (mg) |
Swelling index (%) |
| C1 | 3.8± 0.63 | 10± 0.00 | 5.1± 0.22 | 0.19± 0.22 | 350.7±0.12 | 17.48± 0.22 | 54.2± 0.63 |
| C2 | 4.5±0.07 | 10± 0.00 | 5.3±0.15 | 0.17± 0.15 | 399.2± 0.17 | 17.68± 0.15 | 58.3±0.07 |
| C3 | 5±0.05 | 10± 0.00 | 5.2±0.12 | 0.24± 0.12 | 450.9±0.13 | 18.05± 0.12 | 71.4± 0.05 |
| C4 | 5.3±0.87 | 10± 0.00 | 5.3±0.18 | 0.15±0.18 | 500.6± 0.44 | 18.34± 0.18 | 80.6± 0.87 |
| C5 | 3.7±0.73 | 10± 0.00 | 5.1±0.22 | 0.28± 0.22 | 349.5± 0.48 | 19.88± 0.22 | 58.8± 0.73 |
| C6 | 4.8±0.37 | 10± 0.00 | 5.0±0.05 | 0.42± 0.05 | 397.5± 0.53 | 17.71± 0.05 | 60.6± 0.37 |
| C7 | 5.1±0.56 | 10± 0.00 | 5.1±0.17 | 0.46± 0.17 | 449.9±0.31 | 17.91± 0.17 | 68.3± 0.56 |
| C8 | 5.3±0.57 | 10± 0.00 | 5.2±0.19 | 0.32± 0.19 | 499.4± 0.28 | 18.68± 0.19 | 98.5± 0.57 |
| C9 | 3.9±0.53 | 10± 0.00 | 5.3±0.17 | 0.74± 0.17 | 350.3±0.24 | 18.51± 0.17 | 54.2± 0.53 |
| C10 | 4.5± 0.07 | 10± 0.00 | 5.4± 0.22 | 0.87± 0.22 | 400.1± 0.12 | 19.28± 0.22 | 66.8± 0.07 |
| C11 | 5±0.57 | 10± 0.00 | 5.5±0.15 | 0.75± 0.15 | 449.7± 0.17 | 18.74± 0.14 | 71.6± 0.57 |
| C12 | 5.4±0.97 | 10± 0.00 | 5.1±0.12 | 0.17± 0.12 | 500.1± 0.13 | 17.94± 0.13 | 98.1± 0.97 |
| C13 | 4.1±0.07 | 10± 0.00 | 5.8±0.18 | 0.82± 0.18 | 349.4± 0.44 | 18.22± 0.11 | 57.3±0.07 |
| C14 | 5.3±0.63 | 10± 0.00 | 5.8±0.22 | 0.27± 0.22 | 401.3± 0.48 | 18.97± 0.20 | 70.8± 0.63 |
| C15 | 5.8±0.99 | 10± 0.00 | 5.7±0.05 | 0.66± 0.05 | 449.3± 0.53 | 19.37± 0.05 | 72.0± 0.99 |
| C16 | 5.8±0.99 | 10± 0.00 | 5.8±0.17 | 0.70± 0.17 | 499.3± 0.31 | 19.31± 0.18 | 93.8± 0.99 |
ᴪAll values are expressed as mean± SD, n=5*/10**/20***
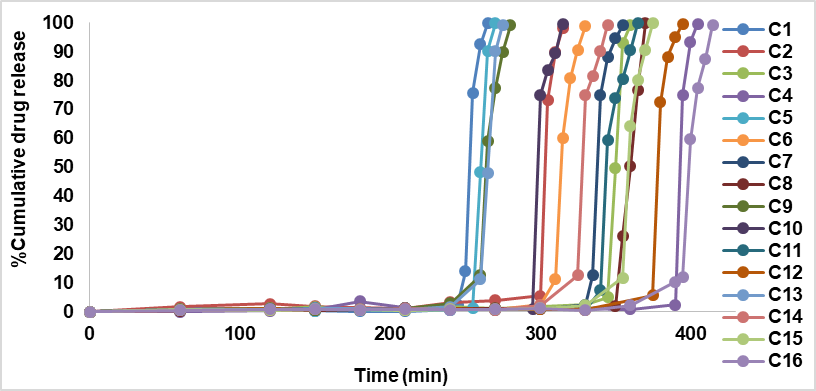
Fig. 2: In vitro release profile of compression-coated tablets (C1 to C16)
Kinetic study
The value of release exponent (n) was found to be a function of the polymer used and the physicochemical property of a drug molecule itself. When the data was plotted as per zero-order kinetics, plots were obtained with correlation coefficient values ranging from 0.2928-0.4422. First-order plots showed correlation coefficient values ranging from 0.2693-0.5930. From the observations, it was concluded that the selected formulations followed first-order release indicating that the dissolution rate of the drug was dependent on the amount of drug available for dissolution. When the drug release data was fitted to the Higuchi equation, correlation coefficient values ranging from 0.1916-0.2944 were obtained. The drug release was proportional to the square root of time, indicating that the drug release from all the compression-coated tablets was time-controlled. The n values of different compression-coated tablets were found in the range of 0.3672–0.5904 in the Korsemayer-Peppas model. The above observations led to the conclusion that all the compression-coated tablets followed time-controlled non-fickian anomalous first-order kinetics. The results are given in table 7.
Table 7: Kinetic profile of compression-coated tablets (C1–C16)
| Formulation code | Zero-order | First order | Higuchi plot | Korsmeyerpeppas plot |
| r2 | r2 | r2 | n | |
| C1 | 0.292 | 0.269 | 0.191 | 0.387 |
| C2 | 0.387 | 0.553 | 0.253 | 0.510 |
| C3 | 0.313 | 0.543 | 0.205 | 0.483 |
| C4 | 0.338 | 0.490 | 0.221 | 0.525 |
| C5 | 0.358 | 0.293 | 0.203 | 0.367 |
| C6 | 0.391 | 0.503 | 0.224 | 0.526 |
| C7 | 0.380 | 0.488 | 0.217 | 0.524 |
| C8 | 0.322 | 0.501 | 0.213 | 0.470 |
| C9 | 0.442 | 0.593 | 0.293 | 0.590 |
| C10 | 0.412 | 0.414 | 0.272 | 0.491 |
| C11 | 0.415 | 0.489 | 0.281 | 0.554 |
| C12 | 0.441 | 0.497 | 0.294 | 0.507 |
| C13 | 0.362 | 0.515 | 0.235 | 0.495 |
| C14 | 0.439 | 0.589 | 0.292 | 0.557 |
| C15 | 0.421 | 0.560 | 0.280 | 0.537 |
| C16 | 0.308 | 0.555 | 0.257 | 0.465 |
*Average of 3 determinations
FTIR studies
The detailed study of the IR spectra of the drug and formulation suggested that the drug is in pure form and all the peaks are in accordance with the standard IR of the pure sample of drug. The IR spectra of formulation C4 (HPMC K4M), C8 (HPMC K15M), C12 (HPMC K100M) and C16 (HPC) exhibited characteristic absorption bands in the corresponding IR regions and results. It is clear from the results that in prepared formulations the difference in the values of characteristic absorption bands indicating the positions of functional groups and bonds present in the drug molecule is negligible and is well within the permissible range. Thus, it can be said that the spectra of drug and formulations are almost identical, suggesting that the drug remains in the same normal form before and after its use in the preparation of formulations. From the above discussion, it can be concluded that no interaction was observed between the drug and various types of polymers used to preparae different formulations. The results of FTIR studies are shown in fig. 3-6.
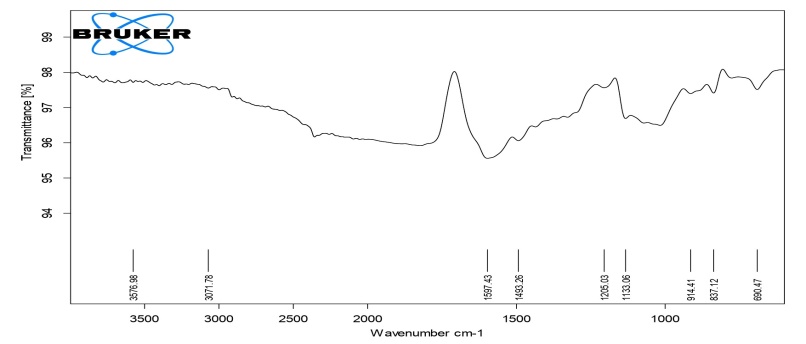
Fig. 3: FTIR Spectra of compression-coated tablet C4 (HPMC K4)
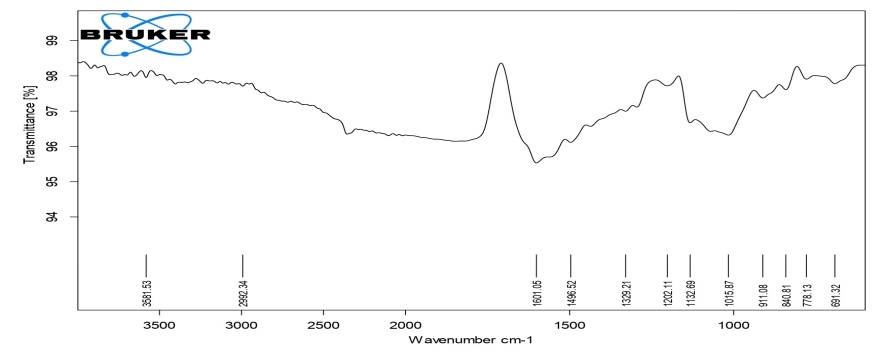
Fig. 4: FTIR Spectra of compression-coated tablet C8 (HPMC K15)
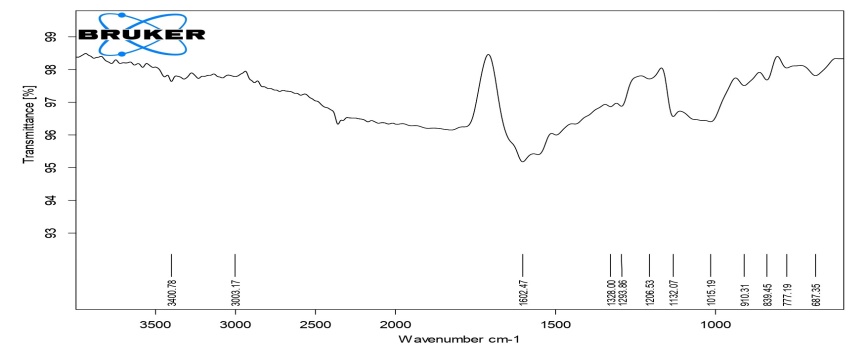
Fig. 5: FTIR spectra of compression-coated tablet C12 (HPMC K100)
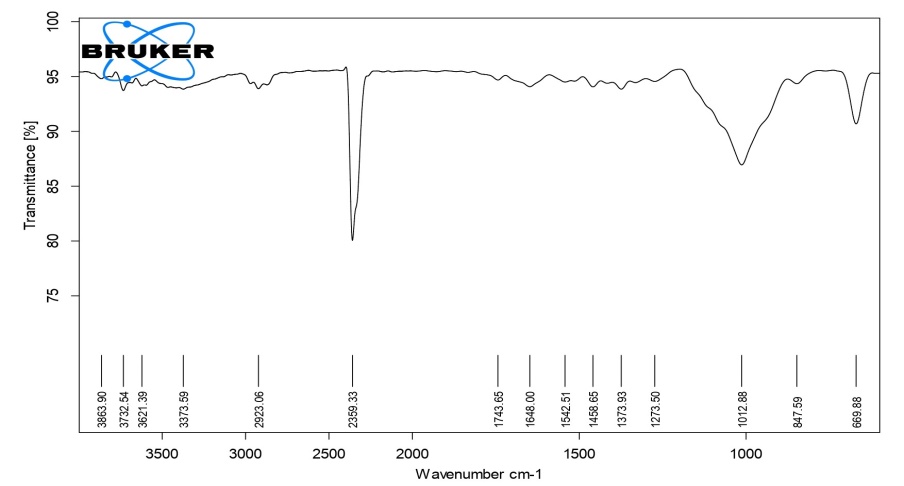
Fig. 6: FTIR spectra of compression-coated tablet C16 (HPC)
CONCLUSION
The research investigates the formulation and characterization of rabeprazole sodium compression-coated tablets utilizing super disintegrants and various polymers through a direct compression method. Preformulation studies affirmed the compatibility of rabeprazole sodium with the selected excipients. Core tablets demonstrated favourable micromeritic properties and were fabricated with sodium starch glycolate, crospovidone, and croscarmellose sodium, exhibiting significant drug release rates ranging from 70.20% to 99.62%. Compression-coated tablets utilizing HPMC K4M, HPMC K15M, HPMC K100M, and HPC were successfully produced, with optimized formulation C5 (HPMC K15M) achieving a lag time of 4 h 15 min and a drug release of 99.85% within 15 min post-lag. The swelling index metrics highlighted HPMC K15M's superior performance, and the release kinetics were identified as non-anomalous first order. Overall, the study demonstrated that rabeprazole sodium compression-coated tablets can be effectively developed for chronotherapeutic applications, and further in vivo evaluations can be performed in the future.
ACKNOWLEDGEMENT
The authors are thankful to K. Somasekhar, Head–Quality Assurance, Enal Drugs Private Limited-Jeedimetla Hyderabad, Telangana, for providing the gift sample of drug Rabeprazole sodium to carry out the research work. The authors are thankful to Dr. R. H. Udupi, NET Pharmacy College, Raichur for providing help to carry out this research.
FUNDING
Nil
AUTHORS CONTRIBUTIONS
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Vijay Archak and H. Doddayya. Sarfaraz Md interpreted the results and was a major contributor in writing the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTERESTS
Declared none
REFERENCES
Upendra K, Sampada HC, Hariprasanna RC. Pulsatile drug delivery systems: current scenario. J Pharm Sci Bio-Sci Res. 2013 Jan 1;3(1):35-8.
Langtry HD, Markham AH. Rabeprazole: a review of its use in acid-related gastrointestinal disorders. Drugs. 1999 Oct;58(4):725-42. doi: 10.2165/00003495-199958040-00014, PMID 10551440.
Badve SS, Sher P, Korde A, Pawar AP. Development of hollow/porous calcium pectinate beads for floating-pulsatile drug delivery. Eur J Pharm Biopharm. 2007 Jan;65(1):85-93. doi: 10.1016/j.ejpb.2006.07.010, PMID 16971097.
Sawada T, Kondo H, Nakashima H, Sako K, Hayashi M. Time-release compression-coated core tablet containing nifedipine for chronopharmacotherapy. Int J Pharm. 2004 Aug 6;280(1-2):103-11. doi: 10.1016/j.ijpharm.2004.05.004, PMID 15265551.
Ueda S, Hata T, Asakura S, Yamaguchi H, Kotani M, Ueda Y. Development of a novel drug release system, time-controlled explosion system (TES). I. Concept and design. J Drug Target. 1994;2(1):35-44. doi: 10.3109/10611869409015891, PMID 8069582.
Sharma SN, Sonawane RS. Role of superdisintegrants in immediate release tablets: a review. Journal of Pharmaceutical and BioSciences. 2017 May 1;5(1):1-5. doi: 10.31555/jpbs/2017/5/1/1-5.
Saitoh T, Watanabe Y, Kubo Y, Shinagawa M, Otsuka K, Ohkawa SI. Effect of H2 blockers on the circadian rhythm of intragastric acidity. Biomed Pharmacother. 2002 Nov;56(2)Suppl 2:349s-52s. doi: 10.1016/s0753-3322(02)00315-3, PMID 12653192.
Kanaka DD, Narasimha Rao N, Sai Mrudula B, Abhinaya M. An investigation and comparison of natural polymers as barrier layers in predictable pulsatile drug release. Drug Discov. 2013 Jan 16;3(7):7-12.
Kanaka DD, Sai Mrudula B, Prameela Rani A. Chronomodulated drug delivery system of montelukast sodium. Pharm Lett. 2010;2(5):316-29.
Kala NP, Shastri DH, Shelat PK. Design and characterization of buccoadhesive liquisolid system of an antihypertensive drug. J Drug Deliv. 2015;2015:574247. doi: 10.1155/2015/574247, PMID 26579235.
Unites States Pharmacopoeia Convention. United States pharmacopoeia 38-national formulary. Vol. 33. Stationery Office; 2010.
Lachman L, Lieberman HA, Kanig JL. The theory and practice of industrial pharmacy. 2nd ed. Philadelphia: Lea & Febiger; 1976.
Senthikumar K, Muthukumaran M, Chenchuratnam B. Formulation and evaluation of rabeprazole sodium enteric coated pellets. Int J Adv Pharm Biol Chem. 2012 Jan-Mar;1(1):7-14.
Kwabena OK, Frederic OY, Samuel LK. Formulation and quality evaluation of two conventional release tablet formulations. Int J Pharm Sci Rev Res. 2010 Sep-Oct;4(1):94-9.
Preeti VA, Agarwal V, Agarwal A. An overview on mouth dissolving tablet: from manufacturing and patented technique to quality control test. Asian J Pharm Clin Res. 2022;15(11):7-13. doi: 10.22159/ajpcr.2022.v15i11.46555.
Saddam JN, Laith HS, Mowafaq MG. Preparation and evaluation of oral disintegrating tablets of ketoprofen by direct compression. Iraqi J Pharm Sci. 2012;21(2):63-8.
Bardonnet PL, Faivre V, Pugh WJ, Piffaretti JC, Falson F. Gastroretentive dosage forms: overview and the special case of Helicobacter pylori. J Control Release. 2006 Mar 10;111(1-2):1-18. doi: 10.1016/j.jconrel.2005.10.031, PMID 16403588.
United States Pharmacopeia and National Formulary (USP 34-NF 29). Rockville, MD: United States Pharmacopeia convention; 2011.
Reza MS, Quadir MA, Haider SS. Comparative evaluation of plastic, hydrophobic and hydrophilic polymers as matrices for controlled-release drug delivery. J Pharm Pharm Sci. 2003 May-Aug;6(2):282-91. PMID 12935440.
Chella N, Shastri NR, Tadikonda RR. Use of the liquisolid compact technique for improvement of the dissolution rate of valsartan. Acta Pharm Sin B. 2012 Oct;2(5):502-8. doi: 10.1016/j.apsb.2012.07.005.
Mahale Nitin B, Battase AP. Formulation development and evaluation of orodispersible tablets of valsartan using spray-dried diluents. Indo Am J Pharm Res. 2018;8(10):1012-27.
Sanjiv KG, Sunil K. Formulation and evaluation of fast dissolving tablet of nimodipine using different super disintegrants. World J Pharm Pharm Sci. 2020;9(4):531-52.
Saripilli R, Yerni KM. Formulation and evaluation of famotidine fast-dissolving tablets using synthetic superdisintegrants. Int J Pharm Pharm Sci. 2019;11(8):17-25.
Jagdale SC, Suryawanshi VM, Pandya SV, Kuchekar BS, Chabukswar AR. Development of press-coated, floating-pulsatile drug delivery of lisinopril. Sci Pharm. 2014 Apr 14;82(2):423-40. doi: 10.3797/scipharm.1301-27, PMID 24959410.