
Int J Pharm Pharm Sci, Vol 18, Issue 2, 5-12Original Article
NOVEL MEFENAMIC ACID-ANTIBIOTIC COMBINATIONS AS POTENTIAL INHIBITORS OF SARS-COV-2 MAIN PROTEASE
DIVYA PINGILI, SOWJANYA V., ARCHANA AWASTHI*
Department of Pharmaceutical Chemistry, Sri Venkateshwara College of Pharmacy, Madhapur, Hyderabad-500081, Telangana, India
*Corresponding author: Archana Awasthi; *Email: archi.rinki@gmail.com
Received: 06 Oct 2025, Revised and Accepted: 24 Dec 2025
ABSTRAC
Objective: The ongoing search for effective COVID-19 treatments has driven interest in drug repurposing and hybrid drug design strategies. This study aimed to develop and evaluate novel mefenamic acid–antibiotic hybrids as potential inhibitors of the SARS-CoV-2 main protease (Mpro).
Methods: Eight hybrid molecules were rationally designed and analyzed using molecular docking and in silico ADME evaluations to predict their binding affinity, stability, pharmacokinetic, and pharmacodynamic behaviour.
Results: Eight mefenamic acid–antibiotic hybrids were evaluated for their potential to inhibit SARS-CoV-2 Mpro. All hybrids demonstrated stronger binding affinity than mefenamic acid (MEF) alone, with the MEF–cephalexin (MEF–CEX) conjugate showing the most favorable binding energy (–7.6 kcal/mol), indicating enhanced complex stability due to the cephalosporin scaffold. ADME predictions revealed moderate pharmacokinetic properties across the series, and the MEF–ciprofloxacin (MEF–CIP) hybrid displayed notable blood–brain barrier permeability despite one Lipinski rule violation.
Conclusion: The findings highlight mefenamic acid–antibiotic hybrids, particularly cephalosporin-based derivatives, as promising multifunctional candidates with combined anti-inflammatory and antiviral potential against SARS-CoV-2. Further preclinical validation is warranted to optimize their pharmacological and safety profiles for potential therapeutic application.
Keywords: COVID-19, Mefenamic acid, Antibiotics, Molecular docking, ADME properties, Main protease (Mpro)
© 2026 The Authors. Published by Innovare Academic Sciences Pvt Ltd. This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/)
DOI: https://dx.doi.org/10.22159/ijpps.2026v18i2.57105 Journal homepage: https://innovareacademics.in/journals/index.php/ijpps
INTRODUCTION
COVID-19, first reported in Wuhan, China, in 2019, is caused by SARS-CoV-2, a betacoronavirus belonging to the Sarbecovirus subgenus of the Coronaviridae family. The viral particle is composed of four key structural proteins: spike, membrane, nucleocapsid, and envelope (fig. 1). Despite the global urgency, no gold-standard antiviral therapy for COVID-19 currently exists, contributing to high infection and mortality rates worldwide [1].
The traditional drug development pipeline typically requires 10–12 y from proof of concept to market entry, with innovative therapeutic strategies potentially extending up to 15 y. Moreover, the cost of de novo drug discovery is estimated at over $1 billion. Given these challenges, drug repurposing offers a practical alternative by accelerating availability, reducing development timelines, and lowering manufacturing costs [2]. This approach provides a promising means to identify selective and effective antivirals against COVID-19 while easing the burden on healthcare systems [3].
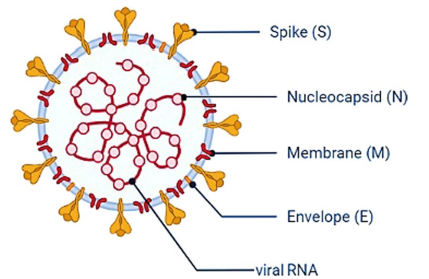
Fig. 1: Structure of SARS-CoV-2 [6]
SARS-CoV-2 encodes a number of proteins, but the primary protease Mpro is crucial to viral replication because it cleaves the larger viral polyproteins into usable non-structural proteins that are needed to construct the replication–transcription complex [4]. The absence of closely similar host analogs and highly conserved substrate selectivity makes Mpro one of the most desirable targets for antiviral drugs [5]. The replicative enzymes, like as Mpro, provide especially intriguing pathways for broad-spectrum antiviral therapy. Spike glycoprotein also well-known role in promoting viral entry through ACE2 binding [6]. The trimeric class I transmembrane glycoprotein enhances viral entry. It is found in the glycoproteins of HCV, HIV, influenza, paramyxovirus, and Ebola virus. SARS-CoV-2 S protein recognises receptors, attaches to cells, and merges with them during infection [7, 8].
During viral replication, the SARS-CoV-2 Mpro, a highly conserved cysteine hydrolase in coronaviruses, plays a crucial role by cleaving viral polyproteins into multiple functional units required for virus maturation [9]. Each Mpro monomer has a molecular weight of approximately 34.21 kDa and consists of three domains (I, II, and III). Among these, domain III, an α-helical domain, is responsible for initiating dimerization, which is essential for enzymatic activation. The monomeric form of Mpro is only weakly active, whereas the dimeric form serves as the fully functional enzyme with maximum hydrolytic activity [10]. Stable interaction between the N-terminus and C-terminus is critical for maintaining this dimeric structure. Mpro contains 12 cysteine residues, of which Cys85, Cys145, and Cys156 are solvent-exposed; notably, Cys145 acts as the key catalytic residue within the active site [11]. Given its central role in viral replication and pathogenesis, SARS-CoV-2 Mpro represents a highly promising therapeutic target for developing antiviral agents against coronaviruses.
Mefenamic acid (MEF), also known as 2-[N-(2,3-dimethylphenyl) amino] benzoic acid (fig. 2), is an anti-inflammatory, analgesic, and antipyretic NSAID [12]. Its pharmacological effect is facilitated by the inhibition of cyclooxygenase (COX) enzymes, resulting in reduced prostaglandin synthesis. Studies suggest that MEF suppresses the NLRP3 [nucleotide-binding domain (NOD), leucine-rich repeat (LRR), and pyrin domain-containing protein 3] inflammasome and IL-1β secretion by blocking the volume-regulated anion (Cl−) channel, in contrast to other NSAIDs [13]. This inhibition is independent of COX-1-mediated anti-inflammatory actions. MEF, in addition to its analgesic and anti-inflammatory properties, has garnered interest in recent years for potential repurposing in viral and inflammatory conditions. MEF significantly reduced viral replication [14].
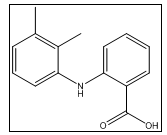
Fig. 2: Structure of Mefenamic acid
The concept of hybrid drug design, combining two bioactive components into a single molecular entity, has gained significant attention for its ability to produce multi-target therapeutic effects [15]. Recent work illustrates the promise of this strategy: Cl-phenylpyrazolone–1,2,3-triazole hybrids synthesized via click chemistry feature a stable ether linkage between pharmacophores and exhibit antiviral activity against coronaviruses [16]. In another example, quinoline–morpholine hybrids developed by incorporating a 1,2,3-triazole ring as a durable linker, further demonstrating the versatility and therapeutic potential of hybrid structures [17]. In this study, we designed eight mefenamic acid–antibiotic hybrid compounds and evaluated their potential as inhibitors of the SARS-CoV-2 main protease (Mpro).
MATERIALS AND METHODS
Rationale for combining mefenamic acid with antibiotics
The combination of MEF and antibiotics is proposed to enhance antiviral efficacy by engaging complementary mechanisms that target both viral and host pathways. The rationale for selecting mefenamic acid, a fenamate-class nonsteroidal anti-inflammatory drug (NSAID), is based on three key considerations. First, its primary mechanism involves cyclooxygenase (COX) inhibition, leading to reduced synthesis of pro-inflammatory prostaglandins. This anti-inflammatory effect may help mitigate the cytokine storm and tissue damage characteristic of severe COVID-19 cases [18, 19]. Second, structural and spectroscopic analyses, including STD-NMR and in silico studies, have identified interactions between MEF and the SARS-CoV-2 Mpro, suggesting a potential inhibitory effect on viral replication [20]. Third, MEF possesses a well-established safety profile, wide availability, and low cost, making it an attractive candidate for drug repurposing. Collectively, these properties indicate that MEF may provide dual benefits: direct anti-inflammatory activity to reduce host-mediated pathology, and potential antiviral or host-targeted effects that complement existing therapies. The angiotensin-converting enzyme 2 (ACE2) receptor functions as a crucial cellular entry point for SARS-CoV-2, effectively acting as the “doorway” that facilitates viral attachment and entry into human cells [21-23]. Although MEF does not directly bind to or inhibit ACE2, its anti-inflammatory and host-modulatory effects may indirectly reduce viral infectivity and disease severity by mitigating inflammatory signaling and modulating host proteases involved in viral entry.
Conversely, antibiotics, apart from their traditional antibacterial roles, have been computationally predicted to interact with key SARS-CoV-2 proteins, including the spike glycoprotein and the Mpro, a pivotal enzyme required for viral replication. Mass spectrometric and biochemical analyses have further demonstrated that certain penicillin derivatives can covalently acylate the active-site cysteine residue (Cys145) of Mpro, confirming a direct inhibitory interaction via enzyme modification [24].
The SARS-CoV-2 Mpro employs a Cys145 within its active site to cleave viral polyproteins into functional subunits essential for replication. This cysteine acts as a nucleophile, rendering Mpro particularly susceptible to electrophilic or acylating agents. In this context, β-lactam antibiotics such as penicillin and cephalosporins are of special interest because their strained four-membered β-lactam ring exhibits high reactivity toward nucleophilic residues. This chemical property allows β-lactams to form covalent acyl–enzyme complexes, the same mechanism through which they inhibit bacterial transpeptidases. Accordingly, β-lactam scaffolds may form analogous covalent bonds with Mpro’s catalytic cysteine, thereby inhibiting viral proteolytic activity and replication [25]. This study also investigates the interaction of the specific fluoroquinolone class of antibiotics with the SARS-CoV-2 Mpro using DFT, molecular docking, and ADME analyses. The results reveal strong binding affinities, suggesting potential inhibitory activity against Mpro [26].
Taken together, these mechanisms suggest that combining MEF with antibiotics could provide a multi-targeted therapeutic strategy simultaneously mitigating viral entry, replication, and inflammatory damage. The proposed multi-mechanistic synergy of mefenamic acid–antibiotic hybrid molecules against SARS-CoV-2 is illustrated in fig. 3.
Design of mefenamic acid-antibiotic hybrid derivatives
The procedure for designing mefenamic acid-antibiotic hybrid derivatives involved a methodical two-step approach to integrate different antibiotics into the structure of MEF effectively. The selection of this technique was made to augment the antiviral effectiveness of MEF by leveraging the therapeutic characteristics of antibiotics. The MEF derivatives are synthesized in two steps. In the first step, chlorine derivatives of MEF were formed and followed by the attachment of various antibiotics to form mefenamic acid-antibiotic derivatives.
Molecular docking
Molecular docking was conducted to evaluate the interactions between mefenamic acid–based ligands and the SARS-CoV-2 Mpro. The crystal structure of SARS-CoV-2 Mpro (PDB ID: 5R7Z) was retrieved from the Protein Data Bank (https://www.rcsb.org/). Ligand structures of MEF and its designed hybrids were created using ChemBioDraw Ultra 14, converted into three-dimensional conformations, and energy-minimized using the Molecular Mechanics 2(MM2) force field until the root mean square (RMS) gradient reached 0.01 kcal/mol·Å. The optimized ligands were saved in PDB format and imported into AutoDock Tools, where Gasteiger partial charges were assigned and all rotatable bonds were defined.
The receptor protein was prepared by removing water molecules, adding polar hydrogens, and converting the structure into PDBQT format using AutoDock Tools. Ligands were further optimized and processed in BIOVIA Discovery Studio. Docking simulations were performed using AutoDock Vina integrated in PyRx, with the docking grid centered on the catalytic dyad residues His41 and Cys145 of Mpro. The grid box parameters were as follows:
Center (x, y, z): –10.58, 12.73, 68.33
Dimensions (Å): 60 × 60 × 60
Grid spacing: 0.375 Å
These coordinates fully encompassed the substrate-binding pocket, including the catalytic site and adjacent subsites. The resulting protein–ligand complexes were visualized and analyzed using BIOVIA Discovery Studio to identify key binding interactions and conformational orientations. Additionally, complementary computational analyses were applied to estimate binding free energy, affinity, and other physicochemical parameters, providing deeper insights into the ligand–target interaction dynamics. This workflow enabled a robust assessment of the binding affinities and interaction profiles of MEF and its hybrid derivatives [27, 28].
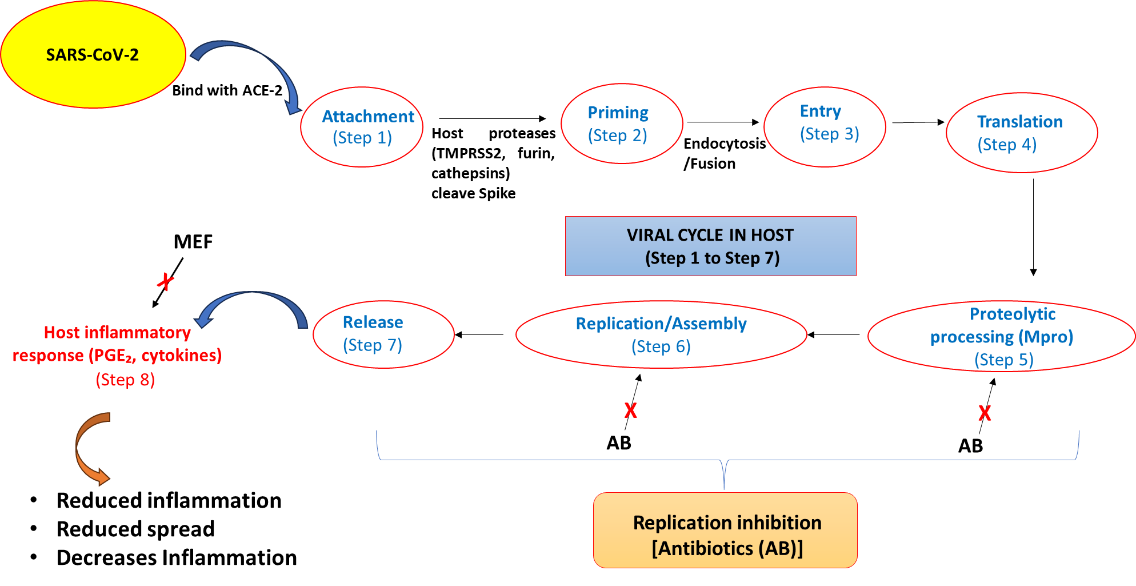
Fig. 3: Combined mechanistic diagram for mefenamic acid-antibiotics synergy against SARS-CoV-2
Table 1: Selected antibiotics for the design of mefenamic acid-antibiotic hybrid derivatives
| Drug | Molecular formula | Structure |
Amoxicillin (β-lactam Antibiotics-Penicillin) |
C16H19N3O5S | 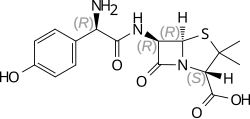 |
Ampicillin (β-lactam Antibiotics-Penicillin) |
C16H19N3O4S | 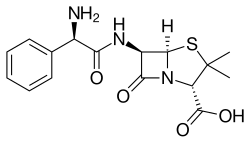 |
Cephalexin (β-lactam Antibiotics-Cephalosporin) |
C16H17N3O4S | 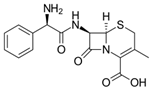 |
Cefaclor (β-lactam Antibiotics-Cephalosporin) |
C15H14ClN3O4S | 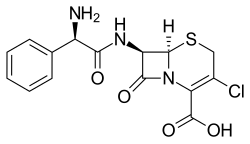 |
Cefadroxil (β-lactam Antibiotics-Cephalosporin) |
C16H17N3O5S | 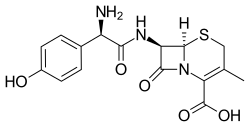 |
Cefuroxime (β-lactam Antibiotics-Cephalosporin) |
C16H16N4O8S | 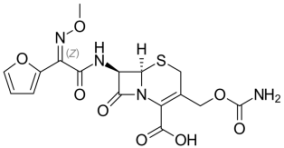 |
Cefotaxime (β-lactam Antibiotics-Cephalosporin) |
C16H17N5O7S2 |  |
Ciprofloxacin (Antibiotics-Fluoroquinolones) |
C17H18FN3O3 | 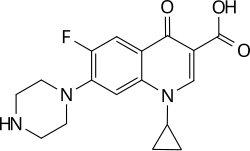 |
In silico ADME properties
ADME studies for all designed mefenamic acid–antibiotic hybrids were carried out using the SwissADME webserver. Drug-likeness was evaluated using Lipinski’s Rule of Five, which predicts the likelihood of good oral bioavailability. According to this rule, most orally active drugs exhibit a molecular weight below 500 g/mol, contain no more than five hydrogen bond donors, and fewer than ten hydrogen bond acceptors, with a logP value not exceeding 5. Compounds that comply with these criteria are considered to have favorable pharmacokinetic properties.
In addition to Lipinski’s rule, other physicochemical parameters such as topological polar surface area (TPSA), solubility (Log S), gastrointestinal (GI) absorption, blood–brain barrier (BBB) permeation, and P-glycoprotein (P-gp) substrate prediction were also assessed. These descriptors provide further insight into absorption, distribution, and central nervous system (CNS) penetration. The combined evaluation of these parameters offers a more comprehensive prediction of the oral bioavailability and drug-likeness of the designed mefenamic acid–antibiotic hybrids [29].
RESULTS AND DISCUSSION
Design of mefenamic acid-antibiotic hybrid derivatives
The mefenamic acid-antibiotic hybrid derivatives were designed based on a stepwise synthesis strategy.
Step 1: Formation of chlorine derivatives of mefenamic acid
The carboxylic acid group is converted into an acid chloride using thionyl chloride. During this reaction, chlorination occurs, replacing the hydroxyl group with a chlorine.
Step 2: Attachment of various antibiotics to the acid chloride derivatives of mefenamic acid
This intermediate then reacts to form an amide bond through the binding of MEF to the free amino group present in various antibiotics (fig. 4). The designed MEF-antibiotic hybrid derivatives are illustrated in fig. 4 and 5.
Hybrid drug design integrating two pharmacologically active components into a single structure, has been identified as a viable technique to produce multi-target action and enhance therapeutic effectiveness.
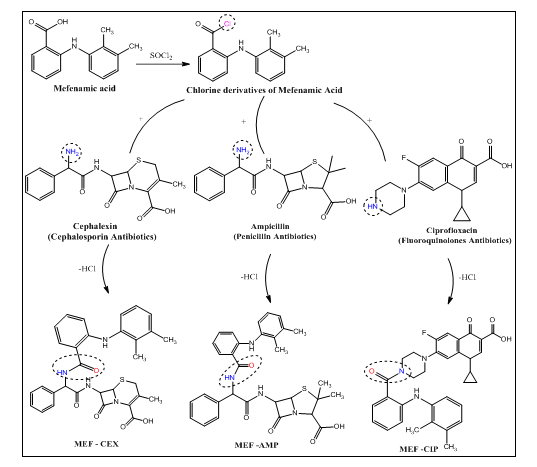
Fig. 4: Designed scheme of mefenamic acid-antibiotic hybrid derivatives
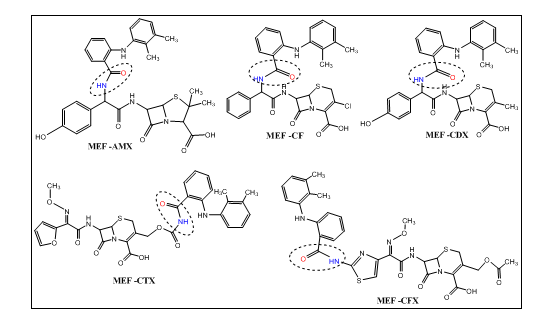
Fig. 5: Designed mefenamic acid-antibiotic hybrid derivatives
Molecular docking
The in silico evaluation of the eight mefenamic acid–antibiotic hybrids revealed enhanced docking affinities toward SARS-CoV-2 Mpro compared with mefenamic acid alone, reinforcing the growing interest in hybrid small-molecule design as a strategy for antiviral discovery. Hybrid small molecules merge two pharmacologically active scaffolds into a single entity, are increasingly recognized for their ability to improve target engagement and enable multi-targeted therapeutic action. Several docking and repurposing studies have reported that cephalosporin scaffolds and certain antibiotics display strong binding to SARS-CoV-2 proteins. Ceftaroline fosamil, a fifth-generation cephalosporin antibiotic, because it contains a 1,2,4-thiadiazole moiety interact with cysteine proteases via its sulfur, making it a candidate for inhibiting viral Mpro [30].
As summarized in table 2, the docking scores for the designed hybrids ranged from –6.0 to –7.6 kcal/mol. MEF alone exhibited a docking score of –6.6 kcal/mol, slightly better than the co-crystallized ligand, indicating its inherent compatibility with the Mpro binding site. Notably, coupling MEF with antibiotic moieties improved binding affinity across all hybrids. The strongest binding was observed for cephalosporin-based hybrids MEF–CEX (–7.6 kcal/mol), MEF–CF (–7.5 kcal/mol), MEF–CTX (–7.3 kcal/mol), and MEF–CDX (–7.2 kcal/mol). This trend suggests that cephalosporin fragments significantly enhance the interaction between the hybrid ligands and key active-site residues of Mpro. Similar findings have been reported in recent studies where hybrid Mpro inhibitors demonstrated improved efficacy through the integration of complementary functional fragments [31].
Table 2: Docking score of the designed derivatives
| Ligand | Code | Binding energy (Kcal/mol) |
| Mefenamic acid | MEF | -6.6 |
| MEF– cephalexin | MEF-CEX | -7.6 |
| MEF– cefaclor | MEF-CF | -7.5 |
| MEF– cefadroxil | MEF-CDX | -7.2 |
| MEF– cefotaxime | MEF-CTX | -7.3 |
| MEF– cefuroxime | MEF-CFX | -7.0 |
| MEF– Ampicillin | MEF-AMP | -6.9 |
| MEF– Amoxicillin | MEF-AMX | -6.8 |
| MEF– ciprofloxacin | MEF-CIP | -6.6 |
| Co – crystallized ligand | CCL | -6.0 |
The docking interaction profiles strongly support the enhanced binding affinity of MEF and its hybrid derivatives. MEF and its conjugates formed a network of stabilizing hydrogen bonds, electrostatic interactions, and hydrophobic/π–π contacts with both catalytic and structural residues of Mpro. The chlorine-substituted aromatic ring of MEF provided a key anchoring point through π–π and alkyl interactions with His 41 and Cys145 (fig. 6a), while additional hydrogen bonds with Glu166, Arg188, and Gln192 further stabilized the complexes.
Fluoroquinolone-based hybrids, such as MEF-CIP, also demonstrated strong interaction profiles, forming hydrogen bonds with Glu166 and Gly143, along with π-alkyl contacts between the piperazine ring and Met49 (fig. 6c). Cephalosporin-derived hybrids, including the MEF–cephalexin conjugate, likewise exhibited robust binding through hydrogen bonding with Glu166 and Thr190 and multiple aromatic interactions involving His41, Met49, Met165, and Pro residues (fig. 6b).
Consistent with prior reports, Glu166 remains the most frequently engaged and functionally critical residue within the protease active site, accompanied by Gln189, His41, and Thr190 [32]. Our molecular docking results confirm that all screened compounds display strong binding affinity (kcal mol⁻¹) toward this catalytic region. The in silico analysis further shows that the hybrid molecules interact with key residues, including Glu166 (via strong hydrogen bonding), Gln189, Gly143, Met 41, His 41, and Thr 190, and occupy the same binding pocket as the reference ligand. This shared interaction pattern highlights their potential as potent Mpro inhibitors.
Collectively, these findings emphasize the dual-interaction capability of MEF–antibiotic hybrids, wherein both hydrogen bonding and hydrophobic contacts contribute to their enhanced affinity. Their ability to engage catalytic and stabilizing residues suggests strong potential to inhibit Mpro activity, thereby suppressing viral replication while retaining the established anti-inflammatory benefits of MEF. Previous literature has already proposed mefenamic acid as a repurposing candidate for COVID-19 due to its anti-inflammatory and possible antiviral actions. The strong binding affinities observed for MEF-based hybrids further reinforce the rationale for designing multifunctional hybrid molecules that integrate anti-inflammatory, antiviral, and antimicrobial properties into a single therapeutic scaffold.
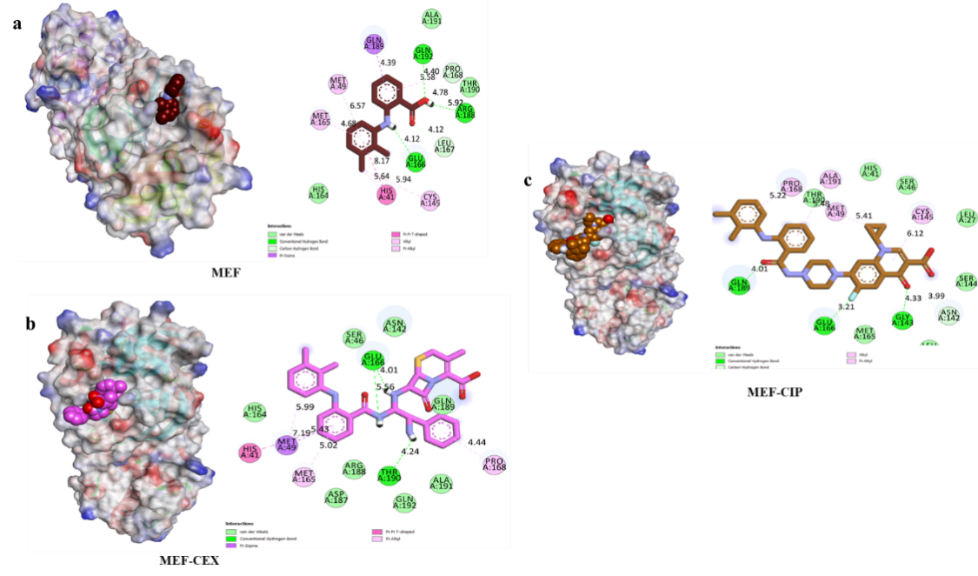
Fig. 6: 2D and 3D interaction diagram of MEF (a), MEF-CEX (b) and MEF-CIP (c) with PDB ID 5R7Z
In silico ADME properties
The physicochemical and pharmacokinetic evaluation of MEF and its hybrid derivatives revealed notable differences driven primarily by the incorporation of bulky β-lactam and fluoroquinolone scaffolds. The physicochemical properties of MEF and its hybrid derivatives with β-lactam antibiotics and ciprofloxacin were assessed in table 3. MEF displayed favorable drug-like characteristics, including low molecular weight (241.29 g/mol), acceptable lipophilicity, good solubility, and full compliance with Lipinski’s rule, supporting its suitability as an orally active compound. In contrast, all hybrid derivatives exhibited substantially higher molecular weights (>550 g/mol), reduced solubility, and a few Lipinski rule violations. These deviations were accompanied by increases in hydrogen bond donors and acceptors as well as markedly elevated polar surface area, particularly in MEF-CTX and MEF-CFX, predicting reduced permeability and diminished oral absorption.
Pharmacokinetic predictions (table 4) further emphasized these trends. MEF demonstrated high gastrointestinal absorption, excellent BBB permeability, and was not identified as a P-gp substrate, highlighting its favorable ADME profile. Conversely, most hybrid derivatives showed poor BBB penetration, low GI absorption, and were predicted to act as P-gp substrates, suggesting that efflux transport may further limit their intracellular availability. Despite these limitations, the fluoroquinolone-based hybrid MEF–CIP emerged as a notable exception. It showed favorable BBB permeability even with one Lipinski violation, consistent with the known CNS penetration of ciprofloxacin. This property suggests potential utility of MEF–CIP in conditions involving CNS-related viral diseases, although further toxicity and safety studies are essential due to the established CNS effects of fluoroquinolones [26].
Overall, the reduced oral drug-likeness observed across many hybrids reflects a recognized challenge in hybrid drug design, where increased molecular size and polarity often improve target binding at the cost of pharmacokinetic performance. However, formulation strategies such as nanoparticle encapsulation, liposomal carriers, lipid–polymer hybrids, and prodrug approaches have shown promise in overcoming these limitations. These delivery systems can enhance solubility, permeability, and bioavailability while mitigating systemic toxicity. Preclinical evidence demonstrating improved antiviral efficacy from nanocarrier-delivered NSAID-antibiotic combinations supports the feasibility of such strategies for MEF-antibiotic hybrids [33]. Collectively, these findings indicate that a hybrid approach increases molecular complexity and enhances binding affinity by introducing additional interaction sites. Optimized formulation approaches will be essential to translate MEF-antibiotic hybrids into viable therapeutic candidates.
Table 3: Physicochemical properties of mefenamic acid–antibiotic hybrid derivatives
| Compounds | M. W (g/mol) | HBD | HBA | TPSA | Log P | Log S | Lipinski's rule of 5 |
| MEF | 241.29 | 2 | 2 | 49.33 | 3.75 | -4.86 | 0 |
| MEF-CEX | 570.66 | 4 | 5 | 153.14 | 3.57 | -6.16 | 1 |
| MEF-CF | 591.08 | 4 | 5 | 153.14 | 3.75 | -6.46 | 1 |
| MEF-CDX | 586.66 | 5 | 6 | 173.37 | 3.28 | -6.26 | 1 |
| MEF-CTX | 647.66 | 4 | 10 | 214.17 | 2.91 | -6.04 | 2 |
| MEF-CFX | 678.74 | 4 | 10 | 242.16 | 2.84 | -5.27 | 2 |
| MEF-AMP | 572.67 | 4 | 5 | 153.14 | 3.44 | -6.44 | 1 |
| MEF-AMX | 588.67 | 5 | 6 | 173.37 | 3.15 | -6.32 | 1 |
| MEF-CIP | 553.62 | 2 | 5 | 89.95 | 5.45 | -7.8 | 1 |
Table 4: Pharmacokinetic properties of mefenamic acid–antibiotic hybrid derivatives
| Compounds | BBB P | GIA | Pgp S |
| MEF | High | Yes | No |
| MEF-CEX | Low | No | Yes |
| MEF-CF | Low | No | Yes |
| MEF-CDX | Low | No | No |
| MEF-CTX | Low | No | Yes |
| MEF-CFX | Low | No | Yes |
| MEF-AMP | Low | No | Yes |
| MEF-AMX | Low | No | Yes |
| MEF-CIP | High | No | Yes |
CONCLUSION
In this study, mefenamic acid–antibiotic hybrids were successfully designed through a streamlined two-step synthetic strategy and evaluated for their antiviral potential using in silico methods. Docking results for the eight hybrid molecules demonstrated improved binding affinities toward SARS-CoV-2 Mpro compared with MEF alone, reinforcing the growing interest in hybrid small molecules and repurposed drug scaffolds as antiviral candidates. Notably, cephalosporin-based hybrids showed the strongest interactions, with the MEF–CEX hybrid exhibiting the most favorable binding energy (–7.6 kcal/mol), suggesting that the cephalosporin moiety significantly enhances Mpro binding.
ADME predictions indicated that although most hybrids showed reduced oral drug-likeness due to their larger and more complex structures, certain candidates, particularly the fluoroquinolone-based MEF–CIP retained desirable properties such as BBB permeability. This highlights the potential of selected hybrids for further exploration in conditions involving CNS-related viral effects, while underscoring the need for comprehensive toxicity and safety assessments.
Overall, the findings align with existing evidence that hybrid molecules can improve target affinity while presenting formulation challenges. This work demonstrates that mefenamic acid can be effectively integrated with antibiotic pharmacophores to generate dual-acting hybrid structures with enhanced interaction toward SARS-CoV-2 Mpro, providing a promising foundation for future antiviral drug development.
ACKNOWLEDGEMENT
The authors are very grateful to Secretary smt. S. Vani Devi, Sri Venkateshwara College of Pharmacy, for providing the essential facilities for the completion of this research work successfully.
ABBREVIATIONS
NSAID: Non-Steroidal Anti-Inflammatory Drugs, MW: Molecular Weight, HBA: Hydrogen Bond Acceptors, Log S: Solubility, HBD: Hydrogen Bond Donors, TPSA: Topological polar surface area, Log P: Lipophilicity, BBB P: Blood Brain Barrier Permeation, GIA: Gastrointestinal absorption, Pgp S: P-glycoprotein substrate.
AUTHORS CONTRIBUTIONS
Idea generation, hypothesis, design of study, and script writing were done by Dr. Divya and Dr. Archana. Dr. Divya implemented and validated the molecular docking scripts. Dr. Divya and Sawjanya performed molecular docking and ADME analysis.
CONFLICT OF INTERESTS
The authors declared no conflict of interest.
REFERENCES
Talele SG, Ahire ED, Surana KR, Sonawane VN, Talele GS. Corona virus disease (COVID-19): a past and present prospective. Asian J Pharm Res. 2022;12(1):45-53. doi: 10.52711/2231-5691.2022.00008.
Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication and pathogenesis. J Med Virol. 2020;92(4):418-23. doi: 10.1002/jmv.25681, PMID 31967327.
Naresh BV. A review of the 2019 novel coronavirus (COVID-19) pandemic. Asian Jour Pharmac Rese. 2020;10(3):233-8. doi: 10.5958/2231-5691.2020.00040.4.
Ullrich S, Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020;30(17):127377. doi: 10.1016/j.bmcl.2020.127377, PMID 32738988.
Goyal B, Goyal D. Targeting the dimerization of the main protease of coronaviruses: a potential broad-spectrum therapeutic strategy. ACS Comb Sci. 2020;22(6):297-305. doi: 10.1021/acscombsci.0c00058, PMID 32402186.
Hu X, Zhou Z, Li F, Xiao Y, Wang Z, Xu J. The study of antiviral drugs targeting SARS-CoV-2 nucleocapsid and spike proteins through large-scale compound repurposing. Heliyon. 2021;7(3):e06387. doi: 10.1016/j.heliyon.2021.e06387, PMID 33688584.
Mousavizadeh L, Ghasemi S. Genotype and phenotype of COVID-19: their roles in pathogenesis. J Microbiol Immunol Infect. 2020;54(2):159-63. doi: 10.1016/j.jmii.2020.03.022, PMID 32265180.
Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587(7835):657-62. doi: 10.1038/s41586-020-2601-5, PMID 32726803.
Hu Q, Xiong Y, Zhu GH, Zhang YN, Zhang YW, Huang P. The SARS-CoV-2 main protease (Mpro): structure function and emerging therapies for COVID-19. MedComm. 2022;3(3):e151. doi: 10.1002/mco2.151, PMID 35845352.
Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289-93. doi: 10.1038/s41586-020-2223-y, PMID 32272481.
Karges J, Kalaj M, Gembicky M, Cohen SM. ReI tricarbonyl complexes as coordinate covalent inhibitors for the SARS-CoV-2 main cysteine protease. Angew Chem Int Ed Engl. 2021;60(19):10716-23. doi: 10.1002/anie.202016768, PMID 33606889.
Moshawih S, Jarrar Q, Bahrin AA, Lim AF, Ming L, Goh HP. Evaluating NSAIDs in SARS-CoV-2: immunomodulatory mechanisms and future therapeutic strategies. Heliyon. 2024;10(3):e25734. doi: 10.1016/j.heliyon.2024.e25734, PMID 38356603.
Daniels MJ, Rivers Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun. 2016;7:12504. doi: 10.1038/ncomms12504, PMID 27509875.
Pareek RP. Use of mefenamic acid as a supportive treatment of COVID-19: a repurposing drug. Int J Sci Res. 2020;9(6):69. doi: 10.21275/SR20530150407.
Navacchia ML, Cinti C, Marchesi E, Perrone D. Insights into SARS-CoV-2: small-molecule hybrids for COVID-19 treatment. Molecules. 2024;29(22):5403. doi: 10.3390/molecules29225403, PMID 39598790.
Musa A, Abulkhair HS, Aljuhani A, Rezki N, Abdelgawad MA, Shalaby K. Phenylpyrazolone-1,2,3-triazole hybrids as potent antiviral agents with promising SARS-CoV-2 main protease inhibition potential. Pharmaceuticals (Basel). 2023;16(3):463. doi: 10.3390/ph16030463, PMID 36986562.
Herrmann L, Hahn F, Wangen C, Marschall M, Tsogoeva SB. Anti-SARS-CoV-2 inhibitory profile of new quinoline compounds in cell culture-based infection models. Chemistry. 2022;28(4):e202103861. doi: 10.1002/chem.202103861, PMID 34859926.
Guzman Esquivel J, Galvan Salazar HR, Guzman Solorzano HP, Cuevas Velazquez AC, Guzman Solorzano JA, Mokay Ramirez KA. Efficacy of the use of mefenamic acid combined with standard medical care vs. standard medical care alone for the treatment of COVID-19: a randomized double-blind placebo-controlled trial. Int J Mol Med. 2022;49(3):29. doi: 10.3892/ijmm.2022.5084, PMID 35029292.
Fazio S, Bellavite P. Early multi-target treatment of mild-to-moderate COVID-19, particularly in terms of non-steroidal anti-inflammatory drugs and indomethacin. BioMed. 2023 Mar 10;3(1):177-94. doi: 10.3390/biomed3010015.
Khan AM, Atia-Tul-Wahab, Farooq S, Ullah A, Choudhary MI. Repurposing of US FDA-approved drugs against SARS-CoV-2 main protease (Mpro) by using STD-NMR spectroscopy, in silico studies and antiviral assays. Int J Biol Macromol. 2023;234:123540. doi: 10.1016/j.ijbiomac.2023.123540, PMID 36740128.
Imam SS, Imam ST, Md Wasif Athar K, Ammar MY. Interaction between ACE2 and SARS-CoV-2 and use of EGCG and theaflavin to treat COVID-19 in initial phases. Int J Curr Pharm Res. 2022;14(2):5-10. doi: 10.22159/ijcpr.2022v14i2.1945.
Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450-4. doi: 10.1038/nature02145, PMID 14647384.
V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19(3):155-70. doi: 10.1038/s41579-020-00468-6, PMID 33116300.
Malla TR, Tumber A, John T, Brewitz L, Strain Damerell C, Owen CD. Mass spectrometry reveals potential of β-lactams as SARS-CoV-2 Mpro inhibitors. Chem Commun (Camb). 2021;57(12):1430-3. doi: 10.1039/D0CC06870E, PMID 33462575.
Malla TR, Brewitz L, Muntean DG, Aslam H, Owen CD, Salah E. Penicillin derivatives inhibit the SARS-CoV-2 main protease by reaction with its nucleophilic cysteine. J Med Chem. 2022;65(11):7682-96. doi: 10.1021/acs.jmedchem.1c02214, PMID 35549342.
Khan MA, Mutahir S, Tariq MA, Almehizia AA. Exploration of specific fluoroquinolone interaction with SARS-CoV-2 main protease (Mpro) to battle COVID-19: DFT, molecular docking, ADME and cardiotoxicity studies. Molecules. 2024;29(19):4721. doi: 10.3390/molecules29194721, PMID 39407649.
Bansal P, Kumar R, Singh J, Dhanda S. In silico molecular docking of SARS-CoV-2 surface proteins with microbial non-ribosomal peptides: identification of potential drugs. J Proteins Proteom. 2021;12(3):177-84. doi: 10.1007/s42485-021-00072-z, PMID 34456530.
Dhanik A, McMurray JS, Kavraki LE. DINC: a new autodock-based protocol for docking large ligands. BMC Struct Biol. 2013;13(Suppl 1):S11. doi: 10.1186/1472-6807-13-S1-S11, PMID 24564952.
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. doi: 10.1038/srep42717, PMID 28256516.
Delgado C, Nogara PA, Miranda MD, Rosa AS, Ferreira VN, Batista LT. In silico and in vitro studies of the approved antibiotic ceftaroline fosamil and its metabolites as inhibitors of SARS-CoV-2 replication. Viruses. 2025;17(4):491. doi: 10.3390/v17040491, PMID 40284934.
Lei S, Chen X, Wu J, Duan X, Men K. Small molecules in the treatment of COVID-19. Signal Transduct Target Ther. 2022;7(1):387. doi: 10.1038/s41392-022-01249-8, PMID 36464706.
Sayed AM, Khattab AR, Aboul Magd AM, Hassan HM, Rateb ME, Zaid H. Nature as a treasure trove of potential anti-SARS-CoV drug leads: a structural/mechanistic rationale. RSC Adv. 2020;10(34):19790-802. doi: 10.1039/D0RA04199H, PMID 35685913.
Abdel Bar HM, Abdallah IA, Fayed MA, Moatasim Y, Mostafa A, El-Behairy MF. Lipid polymer hybrid nanocarriers as a combinatory platform for different anti-SARS-CoV-2 drugs supported by computational studies. RSC Adv. 2021;11(46):28876-91. doi: 10.1039/D1RA04576H, PMID 35478590.